The number of ions belonging to various elements in a sample reveals a particular narrative. In foods and beverages, certain limits for various ions are established to protect health and guarantee a minimum nutrient content. In soil and environmental water sources, heightened levels of specific heavy metal ions can indicate a pollution event has taken place.
In the ultrapure water employed in multiple manufacturing processes, the existence of any ions beyond a certain limit suggests either the source water is unsuitable for use or that the filter system needs maintenance. There are many methods of ion measurement, e.g., ion chromatography (IC), inductively coupled plasma optical emission spectrometry (ICP-OES), or atom absorption spectroscopy (AAS).
All of these are well-proven, broadly applied methods in analytical laboratories. Nevertheless, the preliminary costs are quite high. In comparison, ion measurement employing an ion-selective electrode (ISE) is a promising replacement of these expensive methods. This article describes the challenges which may be faced when using standard addition or direct measurement and how to resolve them to encourage confidence in analysts with this kind of analysis.
Introduction
In 1889, Walther Nernst created the Nernst equation, which encouraged the possibility of the evolution of pH measurement by potentiometric sensors. For approximately 70 years, pH determination was the only analysis in which this equation was of significance. At the start of the 1960s, it was noticed that cuts of lanthanum fluoride crystals demonstrated superb selectivity for fluoride ions1–2.
As such, the first ion-selective electrode next to the pH electrode was constructed. Following this, ion-selective electrodes were slowly cultivated by finding new crystal, glass, gas permeable membrane, and polymer materials. Some of these various ISEs are cited in Table 1.
Table 1. Overview of assorted ion-selective electrodes and their capabilities3.
| Membrane Material |
Ions |
| Crystal |
Ag+, Cu2+, Pb2+, Br-, Cl-, CN-, F-, I-, S2- |
| Polymer |
Ca2+, K+, Na+, Surfactants, NO3- |
| Glass |
Na+, H+ |
| Gas permeable membrane |
NH4+ |
Completing measurements with ion-selective electrodes may sometimes be difficult, but when some crucial points are measured, specific ions of interest can be analyzed easily. In the subsequent sections of this article, general tips for potentiometric ion measurement are offered, in addition to specific ones for basic addition and direct measurement.
General Suggestions
Ion measurement is simple to carry out. The manufacturer’s instructions have to be followed to guarantee both long-term user satisfaction and the precision of the analysis. The subsequent points provide an outline of some common questions and problems
Consequently, it is advised to measure in the linear region to gain accurate and reproducible outcomes. Despite this, an ion concentration measurement in the non-linear (flattened) area by direct measurement is feasible if the sensor is calibrated in this narrow range. Electrodes usually have a measuring range between 10-6 to 1 mol/L.
The measuring range might extend up to 8 mol/L and down to 10-8 mol/L, depending on the sort of ion-selective electrode used. Prior to choosing ion measurement as the selected approach, the analyst needs to be sure that the electrode can measure in the concentration range of the sample.
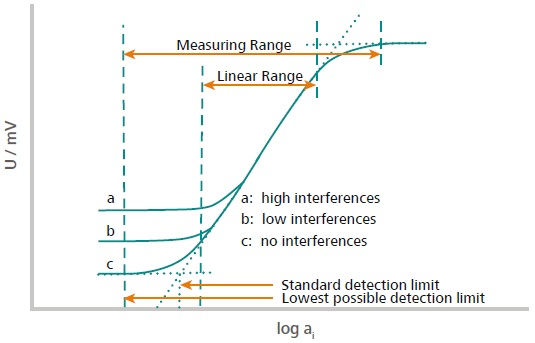
Figure 1. This illustration explains how the measuring range and the linear range are defined. The measuring range is defined from the two points located in the high and low concentration range where no potential change is observed anymore. This means either the concentration is too low, or the sensor is saturated. The linear range is defined as the range where the signal evolves linearly with the activity (concentration) of the ion according to Nernst. Image Credit: Metrohm AG
Interfering Ions
The pH electrode was the first ion-selective electrode created. This electrode displays a superb linear range of 14 decades and is selective for H+. Conversely, most other ion-selective electrodes are not that selective for the measuring ion. There are two distinct kinds of interfering ions: reversible and irreversible. Irreversible interfering ions connect to the membrane material and react with it, or they produce complexes and precipitate with the measuring ion, so it is unavailable for additional analysis.
The presence of the irreversible interfering ion should be avoided if it is reactive towards the membrane; if not, the ISE will be destroyed. Reversible interfering ions exhibit cross-sensitivities because of their comparable size and electronic properties as the measuring ion. These can also bind to the membrane material and contribute to the signal, giving erroneous outcomes. As cross-sensitivities are widespread in ion measurement, the Nernst equation was expanded to the so-called Nikolsky equation:
U = U0 ± ((2.303 × R × T) / (n × F)) × log(ai + Σkij × ajnm)
U: measured potential between ISE and reference electrode in mV
U0: measured potential between ISE and reference electrode at c(ion) = 1 mol/L in mV
R: universal gas constant (R = 8.314 J/(mol∙K))
T: temperature in Kelvin
F: Faraday constant (96485 C/mol)
n: electrical charge of the ion
ai: activity of the ion to be measured
kij: selectivity constant (ion to be measured / interfering ion)
aj: activity of interfering ion
m: electronical charge of the interfering ion
Currently, this equation is employed in most ion meters for direct measurement.
Conditioning of the Electrode
Ion-selective electrodes are usually supplied dry, separate to the membrane material used. To activate the measuring membrane, the electrode must first be conditioned. Conditioning guarantees that the equilibrium of the measuring ion in the membrane is preserved, and as such, an exact measurement is feasible. It is proposed to employ an ion standard solution and a concentration of c(ion) = 0.01 mol/L as the conditioning solution.
Because consistent ion measurements need a steady equilibrium of the measuring ion in the membrane, the ISE must additionally be conditioned in between measurements. In this instance, the conditioning solution is dependent on the concentration of the measuring ion inside the sample. Additional information concerning the appropriate conditioning solution is located in the manufacturer documentation for particular ion-selective electrodes or applications.
Total Ionic Strength Adjustment
The total ionic strength adjustment is necessary because the ion measurement is not dependent on the concentration of the measuring ion in solution but on the activity from then. This parameter differs with the concentration of ions within the solution and additionally with the pH, which impacts the dissociation of certain molecules.
Consequently, as an absolute minimum, a constant ionic background must be maintained, which can be accomplished by adding ionic strength adjuster (ISA). Subject to the measured ion, a fixed pH value is required as well. In that instance, a total ionic strength adjustment buffer (TISAB) is employed.
ISA and TISAB solutions are usually comprised of quite a large concentration of salt, which does not affect the measurement. Suggestions for particular ISA and TISAB solutions are located online or in the manual provided for the ion-selective electrode.
Table 2. Examples of ISA/TISAB solutions for different ionic measurements and information regarding where to find the analysis procedure.
Cleaning of the Electrode
Following every measurement, the electrode is washed fully using deionized water. As they may shorten the lifetime for ion-selective electrodes with crystal membranes and destroy ion-selective electrodes with polymer membranes, organic solvents should not be used for cleaning. When measuring in very small concentration samples, the electrode must be permitted to stand for 10 minutes in deionized water or ISA / TISAB before commencing the next measurement. If not, the ions bound to the electrode membrane material from the former measurement may diffuse out and therefore falsify the signal and the acquired result of subsequent samples.
The deionized water should be swapped prior to every measurement, as diffusion can lead to an improvement in concentration of the measured ion and, consequently, contamination of the deionized water.
Storage of the Electrode
The electrode’s storage requirements depend on the time period of storage and the kind of the electrode (Table 3).
Table 3. Recommendations for the storage of different ISE types.
| Membrane material |
Short storage period |
Long storage period |
| Polymer |
Dry |
Dry |
| Polymer, combined |
In c(ion) = 0.01–0.1 mol/L |
Dry, with some residual moisture |
| Crystal |
In c(ion) = 0.1 mol/L |
Dry, with protective cap |
| Glass |
In c(ion) = 0.1 mol/L |
In deionized water |
Electrode Lifetime
The lifetime of an ISE is dependent on the membrane type, sample matrix, and electrode maintenance. Satisfactory electrode maintenance is comprised of frequent cleaning, appropriate conditioning, and storage, as previously explained in this article. Additionally, it is essential to swap the electrolyte of the reference electrode frequently.
As previously stated, the sample matrix can react with the membrane of the ion-selective electrode. The manual of the electrode typically details substances that should be avoided. For instance, organic solvents may destroy the polymer membrane of ISEs, or acetate will disturb the crystal of the lead ISE.
For ion-selective electrodes with a crystal membrane, the membrane can be regularly revitalized by polishing it. The correct polishing material and technique can usually be located in the ISE manual3. With good upkeep and without lasting sample matrix influences, these kinds of electrodes can endure numerous years of use in the laboratory. The lifetime of polymer membrane electrodes is restricted when compared to those with crystal membranes.
Over time, the polymer membrane ages, causing a loss of performance. This type of ISE typically has a “best before” date, through which they provide their optimal performance. The electrode should be used until this date, and following this, Metrohm can guarantee such performance for a further six months. Typically, the mean lifetime for polymer membrane electrodes with typical laboratory use is around six months. The electrode must be handled carefully; the membrane must not be touched with bare fingers because a deposit of fatty film or sweat from the skin may have a negative impact on the performance of the electrode.
Measuring Modes
Potentiometric ion measurement can be completed in two distinct methods: either by standard addition or by direct measurement. Both of these measuring principles have benefits and drawbacks, which are displayed in Table 4 for ease of comparison. By being aware of the properties of a sample and the frequency of analysis, the correct analysis method may be selected appropriately.
Standard Addition
For ion measurement by standard addition, the sample is first pipetted into a beaker, then ISA/TISAB is added and then numerous increments of standard are also added. Following every addition, the potential is recorded. After this, the ion concentration is calculated iteratively from the prospective difference and volume increment via the method of least squares. Two kinds of standard additions are common. Either specified amounts of standard are inserted into the sample, or the device itself automatically increases the corresponding amount of standard until a difference in potential, which is previously defined, is achieved. The second type of standard addition (defined possible difference) is advised, as it offers more dependable results.
Table 4. Advantages and disadvantages of standard addition and direct measurement.
| Standard addition |
Direct measurement |
| Advantages |
- Matrix independent
- No calibration necessary
- Only a single standard is needed
- Nearly no user interaction necessary
- Electrode is tested with each measurement
|
- Fast measurement (parameters determined quickly, on average in approximately 60 seconds of measuring time)
- Good reproducibility, also at lower concentration ranges
|
| Disadvantages |
- Poor reproducibility in low concentration ranges
- Longer determination times (parameters
- determined within approximately 300 seconds of measuring time)
|
- A series of standards for calibration must be prepared
- Matrix-dependent
- Change of electrode properties is only seen during calibration
|
Ion measurement by standard addition is selected as soon as the matrix is unknown and might include interfering ions that make up part of the measured signal. The total volume of standard added should be between 10 and 90% of the buret volume, and the amount of additions should be four at least (Figure 2). Refilling of the buret must not occur. The additional volume should not surpass 25% of the sample volume, or else dilution will alter the result. Furthermore, there should not be temperature differences between the standard solution and sample solution. The possible difference is also a crucial point to take into consideration. It is intended to add as much standard as necessary to achieve a voltage difference of at minimum 12 mV per addition. The calculation of the outcome is completed iteratively by the device itself.
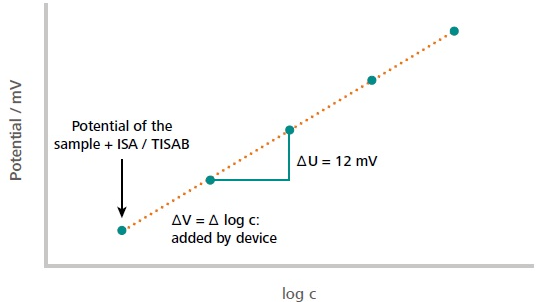
Figure 2. Example of a typical standard addition curve. The first measuring point corresponds to the measured potential of the sample solution (including auxiliary solutions). The subsequent measuring points correspond to the measured potential after each addition of the standard. The initial concentration of the sample is calculated from the measured potential difference in dependence of the added volume increment. The dilution of the sample by auxiliary solutions is considered in the calculations as well. Image Credit: Metrohm AG
If the outcome is not reproducible, the following points must be examined:
- Is the dosing unit tight?
- How much standard is dosed per addition?
- Does the potential measurement between the additions reach a stable potential?
- Is the solution stirred while adding standard?
- Are there air bubbles in the tubing?
- Is the acquired slope within acceptable limits?
Direct Measurement
This kind of ion measurement is done if the sample matrix includes only a small number of interfering substances and is comparable for all samples. Furthermore, samples that include small concentrations of the ion are more appropriate for analysis via direct measurement. These samples create issues with measurement by standard addition: the curve flattens and the evaluation cannot be carried out appropriately (non-linear range).
To ascertain the concentration of the ion in the sample, a calibration must previously be made. The calibration standard concentrations should be selected in such a way that they cover the anticipated concentration of the ion in the sample. The calibration standards are comprised of a specific concentration of the measuring ion in addition to the same proportion of deionized water to ISA/TISAB that is employed following this in the determination itself.
The standards must be measured from the smallest to the largest concentration to lessen the impact of carry over. The analysis is carried out by pipetting the standard into a beaker, adding the corresponding quantity of ISA/ TISAB, and submerging the sensor. The outcome is then shown directly in the chosen concentration unit. Considering the dilution produced by adding ISA/TISAB must not be overlooked.
Should the results be non-reproducible, the following points can be examined:
- Is the same ratio of ISA/TISAB to sample/standard plus water applied for the standard and sample measurements?
- Is the amount of ISA/TISAB equal for each of the standards and for the measuring solution?
- Is the acquired slope of the calibration acceptable?
- Were the standards measured in the correct/defined order?
- Was the electrode properly conditioned between measurements?
- Does the calibration range adequately cover the sample concentration range?
General Points to Consider for Ion Measurement
- Is the standard newly prepared? Some standards can alter their concentration over time.
- What is the age of the electrode? Polymer electrodes in particular age quickly. Metrohm recommends a maximum shelf life of 2 years, dependent on the type of electrode.
- What about the measured value drift? A drift of 1 mV/min at most should be applied.
- Was the sensor appropriately conditioned? If not, the signal and the corresponding results tend to drift.
- The electrode may be tested by immersing the sensor in c(ion) = 1 ∙ 10-4 mol/L and c(ion) = 1 ∙ 10-3 mol/L. If the difference in voltage relates to at least 47.3/z* mV (at 25 °C) (80% of the Nernst Slope), where z* is the charge of the ion, then the electrode is ok and can be used.
Summary
Potentiometric ion measurement by standard addition or by direct measurement is a great option in place of more expensive procedures such as AAS, ICP-OES, or IC. The determination is dependable and quick when respecting the terms of storage, planned use, and measuring range.
References and Further Reading
- Oehme, F. Ionenselektive Elektroden, Grundlagen und Methoden der Direktpotentiometrie; Dr. Alfred Hüthig Verlag GmbH: Heidelberg, 1986.
- Cammann, K. and Galster, H. Das Arbeiten mit ionenselektiven Elektroden - Eine Einführung für Praktiker; Springer-Verlag: Berlin, Heidelberg, New York, 1996.
- Metrohm AG. Ion-selective electrodes (ISE), Metrohm AG: Herisau, Switzerland, 2015. 8.109.8042EN

This information has been sourced, reviewed and adapted from materials provided by Metrohm AG.
For more information on this source, please visit Metrohm AG.