Abstract
The measurement of particle size by dynamic light scattering (DLS) is extremely reliant on how stable the materials being examined are. Contacts across simple, spherical, colloidal particles are often characterized by a balance of attractive and repulsive contacts.
It is these repulsive interactions that lessen the number of collisions between freely diffusing particles, in addition to minimizing the likelihood that a specific collision will cause two surfaces sticking and as such, the outcome will be colloidal stability.
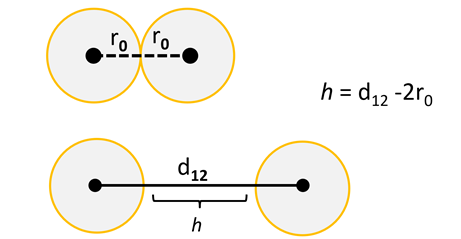
Image Credit: Brookhaven Instrument Corporation
Results
If these interactions are considered in terms of sums of pairwise contacts between two like-charged particles, both of radius ro, distance, h, that describes the surface-to-surface division of colloidal particles can be identified.
The equilibrium of both the attractive and repulsive forces create a complex interaction potential, but the addition of salt is the most direct means to augment this potential. The most immediate impact of salt or ionic strength (I) is to screen these long-range electrostatic repulsions because the most obvious effect of the addition of screening electrolyte is to quash the Debye length, κ-1, or screening constant.
This parameter, often known as the double-layer thickness, directly regulates the distance over which electrostatic impacts falloff, or decay, because there are mobile charge carriers or small ions present. For a 1:1 electrolyte, where I = cs, this can be determined by applying the following expression:
κ-1 = [ϵoϵ kBT/ (2NAe2 cs )] 1/2
Where κ-1 is the Debye screening length. In this expression ϵo and ϵ are the vacuum- and relative- permittivity respectively, kBT is thermal energy expressed in terms of the Boltzmann constant, e is the electronic charge, NA is Avogadro’s number and cs is the molar concentration of salt. This may be simplified at room temperature and conveyed directly in terms of salt concentration:
κ-1 (nm) = 0.301/cs1/2
This permits the calculation of screened electrostatics as an element of surface separation, h. For a given ionic strength, the decay of electrostatic potentials is estimated by the following, where Ψo is the surface potential and κ is the reciprocal of the Debye length (κ-1):
Ψ(h) = Ψo exp(-κh)
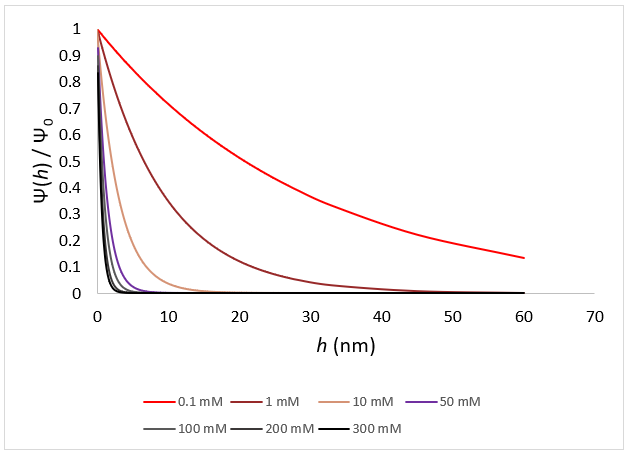
Image Credit: Brookhaven Instrument Corporation
Ionic strength directly regulates the distance over which electrostatics can add to particle stability.
At the smallest salt concentrations, electrostatic repulsions continue for tens of nanometers, as is obvious for salt concentrations <1 mM. At salt concentrations over 50 mM electrostatics have an extremely short range, almost completely falling off at separations larger than several nm. This picture starts to describe the effect of ionic strength on colloidal stability, but yet just focuses on the repulsive element of this interaction. To fully comprehend this, attractive interactions must also be considered.
The most frequently occurring theoretical framework for the comprehension of the stability of colloidal particles is Derjaguin-Landau-Verwey-Overbeek theory (DLVO). DLVO establishes colloidal stability as the sum of van der Waals forces and screened electrostatics (occasionally known as double-layer forces). The form of the interaction potential is complicated; however, at low ionic strength, long-range repulsion is the most dominating. In comparison, at high ionic strength, almost every long-range electrostatic is screened and as such, the attractive element of the potential dominates. When attractive interactions dominate, particles can come closer and have a stronger probability of sticking.
Salt’s Role in Screening Interparticle Interactions
A small quantity of salt can have a large impact on the spaces over which repulsive particle-particle interactions can be felt. As such, it is crucial when preparing samples for DLS or for Zeta Potential Measurements for ionic strength to remain constant. It is uncommon for samples to possess an efficient ionic strength <<0.1 mM, even when not prepared with added salt. Commercially manufactured powders frequently consist of residual salt and buffer. Neutralization of acid or base throughout pH adjustment can also add to the background ionic strength. In practice, it is very challenging to exactly control the total concentration of dissolved ions when working at low salt.
For DLS measurements, it is critical to have the capacity to manage ionic strength, particularly when doing a serial dilution, where variable ionic strength can frequently create an apparent concentration dependence on particle size. Terms like high- and low- salt, although abundant, are subjective; their definitions differ across disciplines.
What are Typical Salt Concentrations?
In biological techniques, ionic strength is frequently formed by dilution in phosphate buffered saline (PBS), which should approximate biological salt and pH, creating an ionic strength of between 120-155 mM differing on the precise structure of the buffer. Nanoparticles are usually resuspended in pure distilled water, which, as previously discussed, is not the same as zero ionic strength and can be altered with ease by trace salt, in addition to adsorption of atmospheric CO2 to form carbonic acid. A further extreme is oil recovery, where it is normal to examine interactions between surfactant micelles in close to brine (>1 M), or brine-like conditions, approaching the solubility limit of simple salts.
Case Study
The salt effect is now measured by the highly charged, semi-flexible polyelectrolyte Sodium Polystyrene Sulfonate (NaPSS). NaPSS is an anionic, or negatively charged, synthetic polymer with large but invariant linear charge density. The sample analyzed below was a commercial polymer with a molecular weight on the order of 1 MDa.
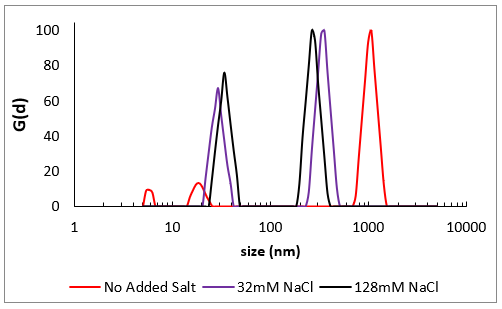
Image Credit: Brookhaven Instrument Corporation
Size distributions from DLS for very high molecular weight NaPSS prepared at three varying ionic strengths. Without added salt (red), repulsions across monomers are not screened and the usually condensed coil-like polymer (black & purple) stretches into a stiff elongated structure, close to the physical length of the polymer chain. At a higher salt level, this impact becomes curbed.
Intra-Particle Interactions and Polyelectrolytes
There are a lot of ways to bring in charge to a molecular surface and all become less energetically advantageous as extremely high charge densities are neared because of charge-charge repulsion. Thermodynamically, it makes it simpler to achieve near the electrolyte or other mobile charge carriers, as these ions lessen the energetic penalty of adding a further charge to a surface that is already highly charged. These direct repulsive interactions can be lessened by colloidal particles by spontaneously maximizing surface-to-surface separation and flexible polymers like polyelectrolytes can reorder themselves structurally to lessen internal repulsions. These long-chain polyelectrolytes lose their semi-compact coil-like structure at the minimum salt concentrations, creating rod-like lengthened structures instead.
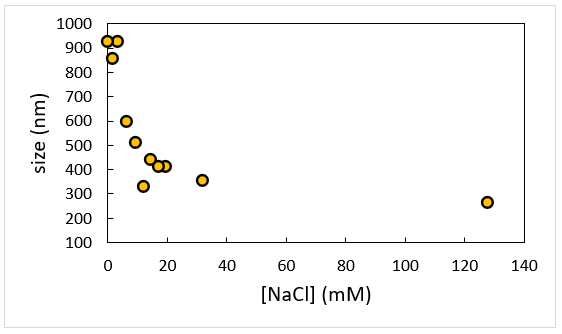
Image Credit: Brookhaven Instrument Corporation
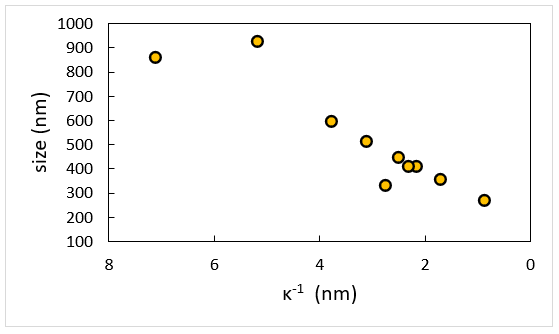
Image Credit: Brookhaven Instrument Corporation
Measurements are taken at constant Cp = 2 g/L NaPSS as a function of ionic strength as defined by the addition of 1:1 electrolyte NaCl. The evident dimensions of polymer chain get large quickly at low-salt as NaPSS gets less flexible and increasingly rod-like. Note that this data is linear when replotted in terms of Debye length, κ-1.
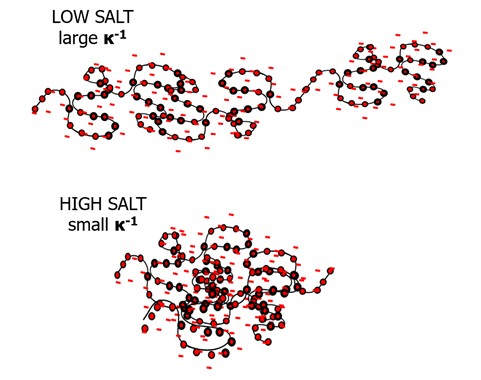
Image Credit: Brookhaven Instrument Corporation
Polyelectrolytes, particularly those with stable, non-titratable charges, get rod-like in the limit of low-, or no-, salt because of repulsion internally occurring between sidechains. This lessening of flexibility at low salt is most obviously seen in the size of the molecule. At minimal ionic strength, a usually compact, flexible polymer starts to rigidify and as such, size measurements show its lengthwise dimension, more so than its group average size. As the impact of salt on polyelectrolyte dimensions can be shown simply using DLS measurements. The degree of rigidity of a polyelectrolyte is extremely proportional to the length-scale over which charged monomers can encounter electrostatic repulsion.
Summary
The balance between long-range repulsion and short-range attraction is known as colloidal stability.
- These electrostatic interactions can be directly modulated by the addition of salt.
- Long-range repulsions are attributed to electrostatics, sometimes known as double-layer forces.
- DLVO provides a good theoretical basis for estimating colloidal stability from first principles.
- Short-range attraction is derived from van der Waals forces.
Ionic strength is unequivocally linked to the strength of small ions in solution.
- There are a lot of definitions of high salt, the consequence of which is industry dependent.
- This is easy for simple monovalent salts and can be more involved for complicated salts or other kinds of small ions, buffers and charged small molecules.
- High salt effectively screens out long-range repulsive electrostatics.
DLS measurements of adaptable charged molecules show this principle.
- Flexible polyelectrolytes get rod-like at low-salt.
Ionic strength impacts are substantial for flexible charged molecules like polyelectrolytes but are not less critical for different kinds of charged surfaces, like those of more conventional colloidal particles. As such, this property is of key importance for colloidal stability and must be considered when preparing samples for light scattering.
References
- Stevens, M.J. and Plimpton, S.J., 1998. The effect of added salt on polyelectrolyte structure. The European Physical Journal B-Condensed Matter and Complex Systems, 2(3), pp.341-345.
- Tadmor, R., Hernandez-Zapata, E., Chen, N., Pincus, P. and Israelachvili, J.N., 2002. Debye length and double-layer forces in polyelectrolyte solutions. Macromolecules, 35(6), pp.2380-2388.
- Lin, M.Y., Lindsay, H., Weitz, D.A., Ball, R.C., Klein, R. and Meakin, P., 1989. Universality in colloid aggregation. Nature, 339(6223), p.360.
- Derjaguin, B.V., Rabinovich, Y.I. and Churaev, N.V., 1978. Direct measurement of molecular forces. Nature, 272(5651), p.313.
- Verwey, E.J.W., Overbeek, J.T.G. and Van Nes, K., 1948. Theory of the stability of lyophobic colloids: the interaction of sol particles having an electric double layer. Elsevier Publishing Company.

This information has been sourced, reviewed and adapted from materials provided by Brookhaven Instrument Corporation.
For more information on this source, please visit Brookhaven Instrument Corporation.