In this interview, AZoM speak with Matt Leclerc, Senior Application Chemist at Nanalysis Corp, about quantitative NMR analysis and how lignins are analyzed using 31P benchtop NMR spectroscopy.
Analysis of Lignins Using 31-P Benchtop NMR Spectroscopy - Nanalysis
Nanalysis XPOD - The Analysis of Lignins Using 31P Benchtop NMR Spectroscopy from AZoNetwork on Vimeo.
Can you introduce the work you do at Nanalysis?
My name is Matt and I am an application chemist at Nanalysis. I will be discussing the analysis of lignins using benchtop NMR spectroscopy. For those who are unfamiliar with our company, Nanalysis manufactures benchtop NMR spectrometers, and we have been a pioneering force in this area for over 10 years. I am very excited to be sharing some of our recent developments with you.
Could you give our readers a little background on NMR technology, and how it applies to spectroscopy?
The history of NMR is very much a history of innovation. From the discovery of the electron’s spin and magnetic moment to Bloch and Purcell’s fundamental discoveries, the old Varian spectrometers a few might remember, to the pioneering work of Bruker who released their 500 MHz spectrometer in the late 70s then their 1000 MHz approximately 30 years later, the field of nuclear magnetic resonance has never stopped evolving.
Finally, since 2010, we have seen the introduction and proliferation of benchtop NMR technology, which has been helping address some of the major accessibility concerns pertaining to the incorporation of traditional high-field spectrometers into many laboratories.
To clarify, I am not saying that benchtop NMR instruments are poised to replace traditional high-field instruments, but rather that they have helped increase the areas in which NMR can be used, allowing more and more people, experts, and beginners alike, to learn about this technique and apply it to their individual workflows.
Could you tell us a little bit about how Nanalysis incorporated NMR technology into its benchtop instruments?
The miniaturization of benchtop NMR technology by Nanalysis was done by:
First – using rare-earth permanent magnets, specifically NdFeB, and arranging them in a highly homogeneous and compact array, which allows us to use minimal amounts of magnetic materials and to have very limited stray fields, meaning the instruments are safe to be around.
The instruments have very little current draws and standard power can be used to power them. They do not require cryogens or significant maintenance, and non-experts can easily use them.
Second - the miniaturization of electronics means that we have been able to condense these down from the large electronics boxes of traditional high-field spectrometers into small circuit board stacks and embed a computer for direct processing if desired. Alternatively, a separate computer can be used to control the instrument.
What types of benchtop NMR spectrometers does Nanalysis offer?
Nanalysis currently offers two main product lines, the 60 MHz and 100 MHz instruments. They are based on NdFeB magnets and have very limited stray fields.
We have a 1H only model for customers doing routine analyses, as well as some more complex heteronuclear experiments available to users hoping to collect additional data from our instruments. All the data I will be talking about today was collected on our 60PRO instrument line.
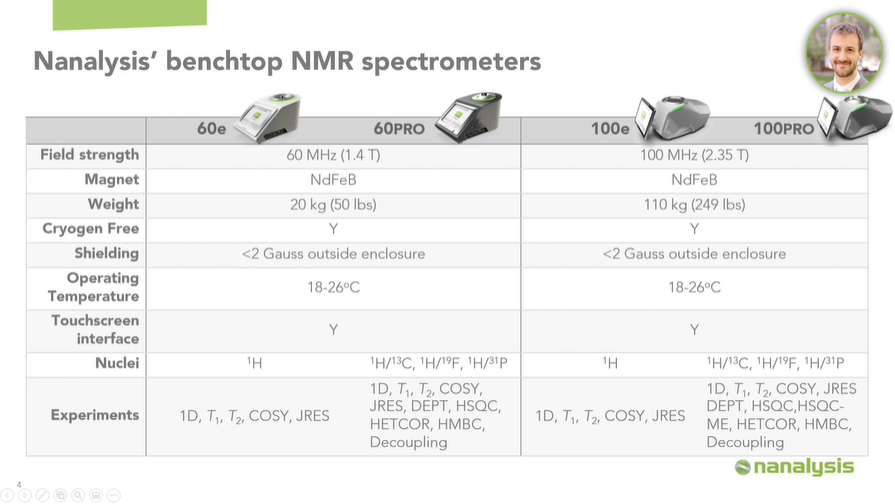
Image Credit: Nanalysis Corp.
How do these benchtop NMR spectrometers compare to traditional high-field spectrometers?
When we think of traditional high-field spectrometers, we know they offer incredible resolution and sensitivity, but they have low accessibility, and it is often not feasible for labs to incorporate these instruments into their workflows, whether it be due to their price, size, or need for expert staff to maintain and use them.
At the other end of the spectrum, we have low-field and low-resolution relaxometers, which are less expensive but can only be used for relatively basic applications.
We like to think of benchtop NMR technology as bridging the gap between these two, by addressing the accessibility issues inherent to high-field instruments, while still providing high-resolution analyses.
There are many underserved markets that could take advantage of the strengths of NMR as a technique, but the combination of price, size requirements and expert staff for maintenance is simply too much for many people to handle, meaning that NMR tends to fly under the radar, no matter how powerful of a technique it is.
Quantitative NMR (qNMR) is rapidly gaining popularity for quantification analysis. Could you tell us a little more about that?
One of the beautiful things about NMR is that it is inherently quantitative, meaning that the integration area of each individual signal is directly proportional to the number of chemically equivalent nuclei giving rise to that signal. This is important because it means that a specific analytical standard of the target analyte is not required.
When we think about HPLC, for example, if you are targeting something like Tadalafil, then you need a specific, certified Tadalafil reference standard to quantify this analyte.
Your instrument needs to be calibrated, and careful consideration must be taken to properly separate all components during a chromatographic run. With NMR, we do not need to separate the components of interest like we do in chromatography, which enables us to obtain a snapshot of everything in solution.
Additionally, a single resonance can be used to quantify the molecule of interest. For example, we were able to quantify Tadalafil using DMT as an internal calibrant. In other words, we used DMT, a compound with a known purity, to determine the purity of Tadalafil.
Tadalafil gives rise to a complex 1H spectrum, with most of the signals overlapping significantly, making it nearly impossible to integrate them accurately. However, since we only need a single resonance to quantify the whole molecule, we were able to obtain great results.
Importantly, the analysis is also non-destructive, meaning that valuable samples can be recovered after analysis.
There are a few main calibrant methods used for qNMR, such as using internal or external calibrants, calibration curves, or even a digital approach called ERETIC.
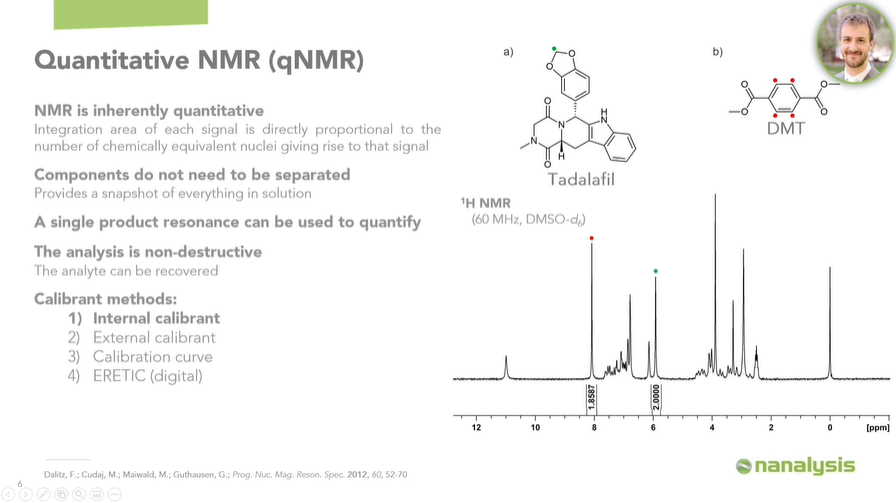
Image Credit: Nanalysis Corp.
How do you use an internal calibrant?
An internal calibrant will typically be weighed directly in the same vial as the analyte of interest and both will be dissolved together and transferred to an NMR tube. For this to be successful, the internal calibrant of choice needs to be of high purity, and this purity must be known.
For example, MilliporeSigma offers a line of TraceCERT® NMR calibrants, which are provided with very accurate purity values. Additionally, the signal of the internal calibrant cannot overlap with other signals in the spectrum, it must be completely soluble in the medium of analysis, and it must not react with anything else in solution.
Using a standard equation, purities and quantities can be readily determined using this approach. This is an extremely powerful technique and one that has been rapidly gaining in popularity over the last several years.
Are there any drawbacks to using qNMR analysis?
As I mentioned, qNMR is rapidly gaining in popularity, but as more people start to use this method, it is important to realize that there are a few key things to consider when preparing for a qNMR analysis.
It is extremely important to be accurate at every step of the way, which means using proper weighing techniques to minimize error (in fact, this is one of the most common sources of error in qNMR). Additionally, proper liquid dispensing methods must be used, which means proper use of a volumetric flask or a calibrated micropipette.
It is crucial that acquisition parameters be optimized for collecting qNMR data. Simplified, this means that an adequate number of points must be used to sample the data, and the repetition time and spectral width must be carefully chosen to enable optimal results.
This cannot be overstated, typical acquisition parameters used to collect traditional 1D NMR data will almost never provide accurate, quantitative results.
Related to the acquisition parameters, scan delays must be chosen such that all spins of interest are fully relaxed between pulses. This is necessary to obtain accurate integrations, which form the basis of qNMR. This is achieved by determining the T1 values of the signals of interest and selecting a scan delay of 5-7 times the longest T1, typically.
Even though the scan delay is technically an acquisition parameter, it remains important and merits to be highlighted: you cannot collect accurate, quantitative data without first selecting the appropriate scan delay.

Image Credit: Nanalysis Corp.
There has been a lot of talk about Heteronuclear NMR. Could you expand on this?
I am sure many have seen 13C experiments, and probably also some 19F and 31P experiments, but it is possible that some of the key advantages of heteronuclear NMR have not been presented to them.
While 1H remains, by far, the most popular nuclide for NMR, it unfortunately has a very narrow chemical shift window, which means that overlap can be, and often is, problematic, even on high-field instruments.
Most of those familiar with even just basic NMR knowledge will be familiar with how the most simple aromatic functionalities can give rise to very complex, often second-order NMR spectra.
Heteronuclides, however, typically have much larger chemical shift ranges, which you can see for lithium-7, fluorine-19, and phosphorus-31, as compared to 1H. This leads to less overlap, even at lower fields, and allows us to differentiate signals that have extremely similar chemical environments.
As a few examples of where heteronuclear NMR is commonly used, 19F is widely used in the pharmaceutical and polymer industries, while 31P is an exceptional handle for organometallic complexes and is commonly used for the analysis of biomolecular samples, such as phospholipids.
Often, heteronuclear NMR is used in conjunction with 1H NMR, but in certain situations, we can gather all the quantitative information we need from heteronuclear NMR alone.
Could you explain why lignin is important?
I do not think I am going to surprise anyone when I say that it is abundantly clear that we need to see widespread use of renewable energy, and we need it soon.
Within this complex framework of renewables, which has become saturated with plenty of buzz words, one of them stands out for the purposes of this presentation – biofuels.
Put simply, biofuels are any fuel derived from biomass, which is essentially any plant material or animal waste.
Within this group, lignin emerges as a class of complex cross-linked phenolic polymers. It is a major component of lignocellulose and is currently made primarily as a by-product of paper production.
Unfortunately, most of the lignin produced around the world continues to be burned for energy production. I used the word unfortunately because one of the wonderful things about lignin is that the monomers which make up its complex structure are important building blocks, which we have historically obtained from petroleum feedstocks.
As I just mentioned, lignin is a cross-linked phenolic polymer, and through the design of some very innovative catalysts over the last decade, we have been able to break down lignin into these fundamental building blocks.

Image Credit: Nanalysis Corp.
Why do you think it is so difficult to study lignin structures?
Well, here is the issue. Determining the structure of lignin is challenging, since it is very complex, and it is different for every single lignin sample. The reason this is an issue is that it is very valuable to know the composition of lignin before attempting to break it down into value-added chemicals.
Using a representative example of a generic lignin structure, the main skeleton making up the structure is formed of repeating units, mostly based on three phenols. In fact, the free radical polymerization of these units is what gives rise to most of the lignin structure.
Even if you are only somewhat familiar with NMR, you will likely take my word for it if I tell you that the 1H and 13C spectra of lignin suffer from significant overlap.
This is the case on both high-field and low-field instruments. If we cannot use 1H or 13C NMR to analyze lignin, at least not easily, and it does not really contain any other useful NMR active nuclei, how can we determine the composition of these polymers?
Thankfully, chemists have derivatized lignin with a wide variety of heteronuclides, the most useful of which has emerged to be 31P. As an NMR handle, 31P is 100% naturally abundant, it has a very large chemical shift range, and it gives rise to sharp lines.
How do you introduce phosphorus into the lignin framework while also maintaining its integrity?
Before proceeding, I would like to mention that the lignin analysis I will be discussing was performed as part of a round-robin study to assess a test method described in a future CSA Group Standard, the CSA Group being the Canadian Standards Association Group. While I cannot divulge the full details of the method quite yet, it is expected to be published in early February.
Additionally, we have also just submitted our own paper summarizing these studies for peer review and are hoping to hear back soon. I would highly encourage anyone interested in lignin analysis to keep an eye out for both publications!
Now, back to the introduction of phosphorus into the lignin framework. We can use a phosphitylation approach, where all the OH groups in the polymer will be transformed according to a general reaction scheme.
Using TMDP as the phosphitylating agent, which is a dioxaphospholane, and cyclohexanol as the internal calibrant, we can identify and quantify all the different substructures which make up lignin.
As a proof of concept, we have phosphitylated several representative model compounds, to illustrate that these will give rise to signals within the 130-150 ppm chemical shift range.
To clarify, this is well-known, but we wanted to confirm these results on our instrument. We have examples showing: an alkyl fragment, a guaiacyl fragment, a carboxylic acid fragment, another alkyl fragment, a basic phenyl fragment, a p-hydroxyphenyl fragment, and finally a syringyl fragment.
There is a peak for the phosphitylated cyclohexanol, around 145 ppm, and the peak for the reaction product between TMDP and trace moisture, which we use as our internal chemical reference at 132.2 ppm. While the lignins are thoroughly dried and moisture is excluded as much as possible during these experiments, we’ll almost always observe a little bit of this product.
To further demonstrate this, we phosphitylated an actual lignin sample, a kraft softwood lignin in this case, and analyzed it on both a benchtop and a high-field instrument. Stacked spectra show that both results look very similar. While the high-field spectrum is slightly better resolved in the aliphatic and carboxylic acid regions, realistically, these two spectra are very alike.
We have demonstrated visually and numerically where the different functionalities arise in the 31P NMR spectrum. The regions have been accurately determined previously, and I encourage anyone to look up some of the references if they would like to know more about them.
The main difference between the hardwood or wheat lignins and the softwood lignins is whether we will be identifying and quantifying 4-O-5’ functionalities, or syringyl functionalities.
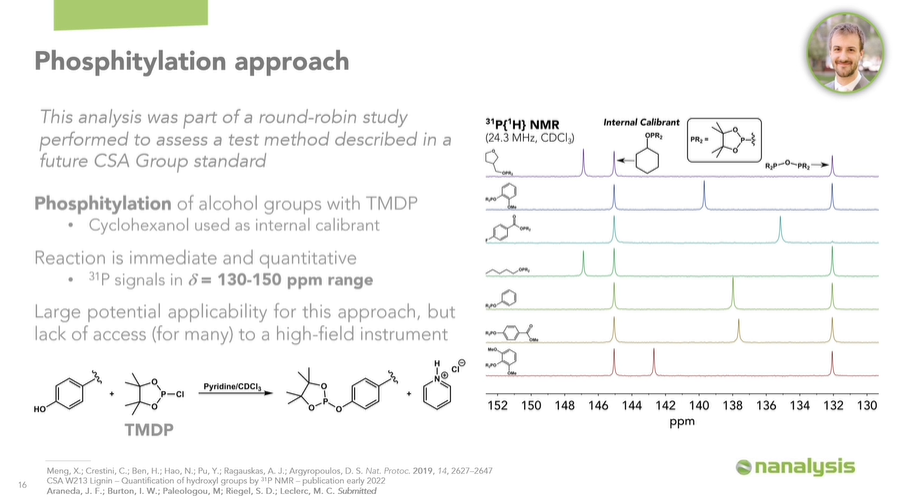
Image Credit: Nanalysis Corp.
What types of lignin did you analyze?
We analyzed two kraft lignins – one softwood, and one hardwood. The 31P spectra from our benchtop instrument were shown. Importantly, we noticed that the syringyl content in the hardwood lignin is much larger than in the softwood lignin, which is exactly what we would expect from numerous previous studies on these types of polymers.
In all cases, the integration area of the internal calibrant was normalized to 1, and the specific integration regions described on the previous slide are used to integrate all the specific substructures of interest.
We next looked at Organosolv hardwood lignin and Soda lignin. Again, we notice some major differences between the two, but primarily the fact that the carboxylic acid content is much higher in the Soda lignin, which is again exactly what we would expect since the Soda pulping process is known to produce these types of lignins.
Additionally, Organosolv lignin has a much larger alkyl content, in the region around 147 ppm.
How does benchtop NMR analysis compare against high-field instruments?
Having confirmed that we could analyze and integrate the various regions of interest in the four types of lignins, we opted to perform a thorough study to determine the performance of our method, especially as it compares to high-field results.
We prepared three samples for each type of lignin and analyzed them all on the benchtop instrument. After processing the spectra, we calculated the concentrations of each substructure, in mmol/g, using the same generic qNMR equation I mentioned earlier.
When we ran these same samples on a high-field instrument we also observed great agreement, which was very encouraging.
This essentially means that we can accurately and reproducibly determine the composition of various lignins, such that anyone working with these products can ascertain what the value of their product might be, so that we can use some of the great catalysts that have been developed to break the most valuable ones down into their monomeric units, instead of obtaining these through petroleum feedstocks.
While the benchtop approach does require additional scans compared to the high-field experiments, it’s important to mention that many laboratories working in these industries don’t have direct access to high-field spectrometers. This, coupled with the fact that the phosphitylated lignins should be analyzed as soon as they’re prepared due to eventual decomposition over time, demonstrates the utility of benchtop NMR technology to help underserved markets gain access to these analyses.
Even though this lignin analysis is extremely promising and could provide value to many of those that produce lignin, it is simply not feasible when samples need to be sent out to a third party for analysis, where samples will sometimes wait in a queue for hours or days.
In conclusion, we analyzed four lignins and quantified their respective substructures using 31P qNMR. We were able to do this by phosphitylating the lignin, transforming all the OH groups into functionalities suitable for 31P NMR. This is important because we cannot use 1H/13C NMR due to the extreme overlapping of resonances.
The results from our 60 MHz instrument matched those from the high-field instrument very closely, and we feel strongly about the fact that while this approach has shown incredible promise over the last several years and has been studied quite a bit, accessibility concerns have made it difficult for the pulp and paper industry to incorporate this method on a large scale.
In addition, the low cost of experiments contributes to making the benchtop NMR approach an attractive path forward for this analysis.
How do you see benchtop NMR spectrometers evolving in the future?
Our vision is one where it becomes possible to develop methods on high-field instruments, like this one for phosphitylated lignin analysis, and implement them at the benchtop level to help industries leverage the power of NMR without many of the requirements of the traditional systems.
One of the key takeaway messages here is that benchtop NMR technology offers unparalleled access to this technique to industries that have historically not been able to afford and use traditional high-field spectrometers. In this sense, the benchtop systems can address accessibility issues for the incorporation of NMR into underserved markets.
About Matt Leclerc Ph.D.
Matt obtained both his BSc in Chemistry and PhD in Organometallic Chemistry at the University of Ottawa. Under the supervision of Prof. R. Tom Baker, his research focused on cobalt fluoroorganometallic catalysis and studying the reactivity of N-heterocyclic carbenes with fluoroalkenes. Following a postdoctoral fellowship with Prof. Warren E. Piers at the University of Calgary, where he explored the synthesis of manganese complexes, he moved into a technical sales role focusing on chromatography and mass spectrometry. Now an application chemist at Nanalysis, he is finally reunited with the technique of NMR, which he used extensively throughout his studies, and is trying to spread the word about the marvels of benchtop technologies!


This information has been sourced, reviewed and adapted from materials provided by Nanalysis Corp.
For more information on this source, please visit Nanalysis Corp.
Disclaimer: The views expressed here are those of the interviewee and do not necessarily represent the views of AZoM.com Limited (T/A) AZoNetwork, the owner and operator of this website. This disclaimer forms part of the Terms and Conditions of use of this website.