Sponsored by PerkinElmerReviewed by Olivia FrostMay 14 2024
Per- and polyfluoroalkyl substances (PFAS) are a class of synthetic compounds discovered in the 1940s commonly used for their surfactant properties.1-3

Image Credit: dba87/Shutterstock.com
Commercial household products, such as nonstick cookware and cleaning supplies, food packaging, water-resistant clothes, and industrial products like automotive lubricants and electronics, are among their many applications.3,4
Because of their broad usage in consumer items and natural persistence, PFAS have leached into the air, soil, and water, resulting in widespread exposure. Several research studies have identified the presence of PFAS in a wide range of matrices (e.g., wastewater, groundwater, drinking water, blood, serum, air, and soil).4-10
Growing concern about the presence and persistence of these compounds in the environment necessitates using commercially available instruments to execute current and evolving regulatory methods simply and reliably.
The United States Environmental Protection Agency has prioritized the development of sensitive, regular, and robust studies for detecting PFAS in diverse matrices, with a primary focus on drinking water. Method 537.1 focuses on the detection of 18 PFAS chemicals in drinking water.11
The EPA later developed a more thorough list, Method 533, which concentrates on additional short-chain PFAS and fluorotelomer alternatives in drinking water.12
In August 2021, the EPA released Draft Method 1633 for analyzing 40 PFAS chemicals in various matrices. As of August 2023, Draft Method 4 has been issued with the finalized multi-laboratory validated study for aqueous sample analysis.13
This article presents validation results for EPA Method 1633 for aqueous matrices utilizing the PerkinElmer LX-50™ UHPLC System coupled with the PerkinElmer QSight® 210 Triple Quadrupole Mass Spectrometry System.
Experimental
Materials and Reagents
Wellington Laboratories provided the primary PFAS standards, extracted internal standards (EIS), and non-extracted internal standards (NIS), as shown in Table 1.
VWR supplied the LC/MS grade water (reagent water) and methanol (MeOH). Formic acid was purchased from Sigma Aldrich. Fisher Scientific supplied the ammonium hydroxide.
Solid phase extraction was performed using a PromoChrom SPE-03 machine equipped with a MOD-004 sample loading device. Phenomenex provided Strata PFAS, polymeric weak anion exchange stacked with graphitized carbon black, and SPE cartridges (200 mg/50 mg, 6 mL).
Sigma Aldrich supplied the 250 mL high-density polyethylene (HDPE) bottles used to prepare and extract all blanks, spiked blanks, field samples, and QC samples.
The HPLC autosampler employed PerkinElmer low-volume 300 µL polyethylene (PE) vials with Sigma Aldrich polyethylene vial caps. Polypropylene vials and caps are necessary to prevent PFAS compounds from adsorbing onto glass vials and to eliminate PFAS materials often found in HPLC vial septa.
Table 1. Target analytes extracted internal standards and non-extracted internal standards. Source: PerkinElmer
| Analyte |
Abbreviation |
| Perfluorobutanoic acid |
PFBA |
| Perfluoropentanoic acid |
PFPeA |
| Perfluorohexanoic acid |
PFHxA |
| Perfluoroheptanoic acid |
PFHpA |
| Perfluorooctanoic acid |
PFOA |
| Perfluorononanoic acid |
PFNA |
| Perfluorodecanoic acid |
PFDA |
| Perfluoroundecanoic acid |
PFUnA |
| Perfluorododecanoic acid |
PFDoA |
| Perfluorotridecanoic acid |
PFTrDA |
| Perfluorotetradecanoic acid |
PFTeDA |
| Perfluorobutanesulfonic acid |
PFBS |
| Perfluoropentansulfonic acid |
PFPeS |
| Perfluorohexanesulfonic acid |
PFHxS |
| Perfluoroheptanesulfonic acid |
PFHpS |
| Perfluorooctanesulfonic acid |
PFOS |
| Perfluorononanesulfonic acid |
PFNS |
| Perfluorodecanesulfonic acid |
PFDS |
| Perfluorododecanesulfonic acid |
PFDoS |
| 1H, 1H, 2H, 2H-Perfluorohexane sulfonic acid |
4:2FTS |
| 1H, 1H, 2H, 2H-Perfluorooctane sulfonic acid |
6:2FTS |
| 1H, 1H, 2H, 2H-Perfluorodecane sulfonic acid |
8:2FTS |
| Perfluorooctanesulfonamide |
PFOSA |
| N-methyl perfluorooctanesulfonamide |
NMeFOSA |
| N-ethyl perfluorooctanesulfonamide |
NEtFOSA |
| N-methyl perfluorooctanesulfonamidoacetic acid |
NMeFOSAA |
| N-ethyl perfluorooctanesulfonamidoacetic acid |
NEtFOSAA |
| N-methyl perfluorooctanesulfonamidoethanol |
NMeFOSE |
| N-ethyl perfluorooctanesulfonamidoethanol |
NEtFOSE |
| Hexafluoropropylene oxide dimer acid |
HFPO-DA |
| 4,8-Dioxa-3H-perfluorononanoic acid |
ADONA |
| Perfluoro-3-methoxypropanoic acid |
PFMPA |
| Perfluoro-4-methoxybutanoic acid |
PFMBA |
| Nonafluoro-3,6-dioxaheptanoic acid |
NFDHA |
| 9-Chlorohexadecafluoro-3-oxanonane-1-sulfonic acid |
9Cl-PF3ONS |
| 11-Chloroeicosafluoro-3-oxaundecane-1-sulfonic acid |
11Cl-PF3OUdS |
| Perfluoro(2-ethoxyethane)sulfonic acid |
PFEESA |
| 3-Perfluoropropyl propanoic acid |
3:3FTCA |
| 2H, 2H, 3H, 3H-Perfluorooctanoic acid |
5:3FTCA |
| 3-Perfluoroheptyl propanoic acid |
7:3FTCA |
| Extracted Internal Standard |
Abbreviation |
| Perfluoro-n-[13C4]butanoic acid |
13C4-PFBA |
| Perfluoro-n-{13C5}pentanoic acid |
13C5-PFPeA |
| Perfluoro-n-[1,2,3,4,6-13C5]hexanoic acid |
13C5-PFHxA |
| Perfluoro-n-[1,2,3,4-13C4]heptanoic acid |
13C4-PFHpA |
| Perfluoro-n-[13C8]octanoic acid |
13C8-PFOA |
| Perfluoro-n-[13C9]nonanoic acid |
13C9-PFNA |
| Perfluoro-n-[1,2,3,4,5,6-13C6]decanoic acid |
13C6-PFDA |
| Perfluoro-n-[1,2,3,4,5,6,7-13C7]undecanoic acid |
13C7-PFUnA |
| Perfluoro-n-[1,2-13C2]dodecanoic acid |
13C2-PFDoA |
| Perfluoro-n-[1,2-13C2]tetradecanoic acid |
13C2-PFTeDA |
| Perfluoro-1-[2,3,4-13C3]butanesulfonic acid |
13C3-PFBS |
| Perfluoro-1-[1,2,3-13C3]hexanesulfonic acid |
13C3-PFHxS |
| Perluoeo-1-[13C8]octanesulfonic acid |
13C8-PFOS |
| Perfluoro-1-[13C8]octanesulfoamide |
13C8-PFOSA |
| N-methyl-d3-perfluoro-1-octanesulfonamidoacetic acid |
D3-NMeFOSAA |
| N-ethyl-d5-perfluoro-1-octanesulfonamidoacetic acid |
D5-NEtFOSAA |
| 1H, 1H, 2H, 2H-Perfluoro-1-[1,2-13C3]hexanesulfonic acid |
13C2-4:2FTS |
| 1H, 1H, 2H, 2H-Perfluoro-1-[1,2-13C3]octanesulfonic acid |
13C2-6:2FTS |
| 1H, 1H, 2H, 2H-Perfluoro-1-[1,2-13C2]decanesulfonic acid |
13C2-8:2FTS |
| Tetrafluoro-2-heptafluoropropoxy-13C3-propanoic acid |
13C3-HFPO-DA |
| N-methyl-d7-perfluorooctanesulfonamidoethanol |
D7-NMeFOSE |
| N-ethyl-d9-perfluoroctanesulfonamidoethanol |
D9-NEtFOSE |
| N-ethyl-d5-perfluoro-1-octanesulfonamide |
D5-NEtFOSA |
| N-methyl-d3-perfluoro-1-octanesulfonamide |
D3-NMeFOSA |
| Nonextracted Internal Standard |
Abbreviation |
| Perfluoro-n-[2,3,4-13C3]butanoic acid |
13C3-PFBA |
| Perfluoro-n-[1,2,3,4-13C4]octanoic acid |
13C4-PFOA |
| Perfluoro-n-[1,2-13C2]decanoic acid |
13C2-PFDA |
| Perfluoro-n-[1,2,3,4-13C4]octanesulfonic acid |
13C4-PFOS |
| Perfluoro-n-[1,2,3,4,5-13C5]nonanoic acid |
13C5-PFNA |
| Perfluoro-n-[1,2-13C2]hexanoic acid |
13C2-PFHxA |
| Perfluoro-1-hexane[18O2]sulfonic acid |
18O2-PFHxS |
Hardware/Software
The analytes were chromatographically separated using a PerkinElmer LX-50 UHPLC System and then detected using a PerkinElmer QSight 210 Triple Quadrupole Mass Spectrometer with electrospray ionization (ESI).
The LX50 Autosampler was altered by substituting all polytetrafluoroethylene (PTFE) based tubing with polyether ether ketone (PEEK) tubing to minimize or remove any PFAS compound contamination caused by the PTFE tubing.
In addition, a PEEK needle was fitted to the autosampler. PerkinElmer Simplicity™ 3Q software was used for all instrument control, acquisition, and processing.
Method
LC Conditions and MS Parameters
Table 2 shows the LC method and MS source parameters. This approach used a pair of C18 columns.
A delay column (PerkinElmer Brownlee™ SPP C18 Column, 50 x 3.0 mm, 2.7 µm) was installed in-line between the LX50 pump and the autosampler to catch and delay potential PFAS interference arising from the LC pump and solvent reservoirs.
To separate PFAS from other interfering components, an analytical column (Brownlee SPP C18 Column, 75 x 4.6 mm, 2.7 µm) and a guard column (Brownlee SPP C18 Column, 5 mm x 4.6 mm, 2.7 µm) were also employed.
The LC gradient program was adjusted from the one specified in EPA Method 1633, as shown in Table 3. The LC program reduces the runtime by two minutes compared to EPA 1633. In addition, the same procedure may be utilized for examination of both 537.1 and 533.14,15
The MS source parameters, which include gas flows, temperature, and position settings, were tuned to achieve optimal sensitivity.
Table 4 shows how the compound-dependent parameters, such as collision energy (CE), entrance voltage (EV), and collision cell lens voltage (CCL2), were optimized for the target compounds. Wherever possible, a secondary mass transition was chosen for confirmation.
Table 2. LC Method and MS Source Conditions. Source: PerkinElmer
| LC Conditions |
| Analytical Column |
Brownlee SPP C18 Column, 75 x 4.6 mm, 2.7 μm (PN: N9308415) |
| Guard Column |
Brownlee SPP C18 Column, 5mm x 4.6 mm, 2.7 μm (PN: N9308532) |
| Delay Column |
Brownlee SPP C18 Column, 50 x 3.0 mm, 2.7 μm (PN: N9308408) |
| Mobile Phase A |
10 mM ammonium acetate in water |
| Mobile Phase B |
Methanol |
| Flow Rate |
0.8 mL/min |
| Column Oven Temperature (°C) |
40 |
| Auto Sampler Temperature (°C) |
15 |
| Injection Volume |
10 |
| Needle Wash 1 |
25% acetonitrile in methanol |
| Needle Wash 2 |
50% water in methanol |
| MS Source Conditions |
| Electrospray Voltage |
-3000 |
| Drying Gas |
110 |
| Nebulizer Gas |
400 |
| Source Temperature (°C) |
200 |
| HSID Temperature (°C) |
280 |
Table 3. LC Gradient Program. Source: PerkinElmer
| Step # |
Time (min) |
Mobile Phase A (%) |
Mobile Phase B (%) |
| 1 |
0.00 |
95 |
5 |
| 2 |
0.70 |
95 |
5 |
| 3 |
1.00 |
55 |
45 |
| 4 |
7.00 |
2 |
98 |
| 5 |
8.00 |
2 |
98 |
| 6 |
8.10 |
95 |
5 |
| 7 |
10.00 |
95 |
5 |
Table 4. Optimized MRM Parameters for the PFAS analytes, extracted internal standards and non-extracted internal standards. Source: PerkinElmer
| Acronym |
Ret Time
(min) |
Precurser
Ion (m/z) |
Product
Ion (m/z) |
CEa |
EVb |
CCL2c |
Quantifier/
Qualifier |
Type |
| 13C4-PFBA |
2.82 |
217.0 |
172.0 |
14 |
-4 |
40 |
Quantifier |
EIS |
| 13C3-PFBA |
2.82 |
216.0 |
172.0 |
14 |
-4 |
40 |
Quantifier |
NIS |
| PFBA |
2.82 |
213.0 |
169.0 |
13 |
-9 |
36 |
Quantifier |
Analyte |
| PFMPA |
3.06 |
229.0 |
85.0 |
30 |
-10 |
70 |
Quantifier |
Analyte |
| 13C5-PFPeA |
3.51 |
268.0 |
223.0 |
15 |
-12 |
45 |
Quantifier |
EIS |
| PFPeA |
3.51 |
263.0 |
219.0 |
15 |
-8 |
80 |
Quantifier |
Analyte |
| 3:3FTCA |
3.56 |
241.0 |
184.0 |
30 |
-3 |
110 |
Quantifier |
Analyte |
| 3:3FTCA |
3.56 |
241.0 |
117.0 |
30 |
-3 |
110 |
Qualifier |
Analyte |
| 13C3-PFBS |
3.6 |
302.0 |
80.0 |
67 |
-28 |
80 |
Quantifier |
EIS |
| 13C3PFBS |
3.6 |
302.0 |
99.0 |
41 |
-28 |
80 |
Qualifier |
EIS |
| PFBS |
3.6 |
299.0 |
80.0 |
65 |
-40 |
240 |
Quantifier |
Analyte |
| PFBS |
3.6 |
299.0 |
99.0 |
40 |
-40 |
240 |
Qualifier |
Analyte |
| PFMBA |
3.73 |
279.0 |
85.0 |
30 |
-10 |
55 |
Quantifier |
Analyte |
| PFMBA |
3.73 |
279.0 |
97.0 |
40 |
-10 |
55 |
Qualifier |
Analyte |
| PFEESA |
3.89 |
315.0 |
135.0 |
30 |
-30 |
75 |
Quantifier |
Analyte |
| PFEESA |
3.89 |
351.0 |
83.0 |
30 |
-30 |
75 |
Qualifier |
Analyte |
| NFDHA |
4.1 |
201.0 |
85.0 |
23 |
-5 |
30 |
Quantifier |
Analyte |
| 13C2-4:2FTS |
4.16 |
329.0 |
81.0 |
53 |
-36 |
60 |
Quantifier |
EIS |
| 13C2-4:2FTS |
4.16 |
329.0 |
309.0 |
29 |
-36 |
60 |
Qualifier |
EIS |
| 4:2FTS |
4.16 |
327.0 |
81.0 |
38 |
-4 |
65 |
Quantifier |
Analyte |
| 4:2FTS |
4.16 |
327.0 |
307.0 |
23 |
-1 |
110 |
Qualifier |
Analyte |
| 13C2-PFHxA |
4.22 |
315.0 |
270.0 |
15 |
-9 |
48 |
Quantifier |
NIS |
| 13C5-PFHxA |
4.22 |
318.0 |
273.0 |
12 |
-4 |
52 |
Quantifier |
EIS |
| PFHxA |
4.22 |
313.0 |
269.0 |
17 |
-10 |
55 |
Quantifier |
Analyte |
| PFHxA |
4.22 |
313.0 |
119.0 |
31 |
-10 |
50 |
Qualifier |
Analyte |
| PFPeS |
4.26 |
349.0 |
80.0 |
73 |
-6 |
100 |
Quantifier |
Analyte |
| PFPeS |
4.26 |
349.0 |
99.0 |
45 |
-6 |
90 |
Qualifier |
Analyte |
| 13C3-HFPO-DA |
4.42 |
287.0 |
169.0 |
12 |
-3 |
44 |
Quantifier |
EIS |
| 13C3-HFPO-DA |
4.42 |
287.0 |
185.0 |
29 |
-3 |
52 |
Qualifier |
EIS |
| HFPO-DA |
4.42 |
285.0 |
169.0 |
10 |
-4 |
70 |
Quantifier |
Analyte |
| HFPO-DA |
4.42 |
285.0 |
185.0 |
38 |
-4 |
70 |
Qualifier |
Analyte |
| 13C4-PFHpA |
4.84 |
367.0 |
322.0 |
17 |
-6 |
75 |
Quantifier |
EIS |
| PFHpA |
4.84 |
363.0 |
319.0 |
16 |
-10 |
56 |
Quantifier |
Analyte |
| PFHpA |
4.84 |
363.0 |
169.0 |
23 |
-10 |
92 |
Qualifier |
Analyte |
| 13C3-PFHxS |
4.86 |
402.0 |
80.0 |
87 |
-8 |
100 |
Quantifier |
EIS |
| 13C3-PFHxS |
4.86 |
402.0 |
99.0 |
40 |
-8 |
100 |
Qualifier |
EIS |
| 18O2-PFHxS |
4.86 |
403.0 |
103.0 |
38 |
-10 |
90 |
Quantifier |
NIS |
| PFHxS |
4.86 |
399.0 |
80.0 |
85 |
-45 |
120 |
Quantifier |
Analyte |
| PFHxS |
4.86 |
399.0 |
99.0 |
49 |
-45 |
87 |
Qualifier |
Analyte |
| ADONA |
4.92 |
377.0 |
251.0 |
17 |
-10 |
62 |
Quantifier |
Analyte |
| ADONA |
4.92 |
377.0 |
85.0 |
64 |
-10 |
62 |
Qualifier |
Analyte |
| 5:3FTCA |
4.99 |
341.0 |
237.0 |
24 |
-10 |
110 |
Quantifier |
Analyte |
| 5:3FTCA |
4.99 |
341.0 |
217.0 |
46 |
-10 |
110 |
Qualifier |
Analyte |
| 13C2-6:2FTS |
5.38 |
429.0 |
409.0 |
33 |
-16 |
124 |
Quantifier |
EIS |
| 13C2-6:2FTS |
5.38 |
429.0 |
81.0 |
43 |
-16 |
124 |
Qualifier |
EIS |
| 6:2FTS |
5.38 |
427.0 |
81.0 |
65 |
-8 |
80 |
Qualifier |
Analyte |
| 6:2FTS |
5.38 |
427.0 |
407.0 |
30 |
-8 |
30 |
Qualifier |
Analyte |
| PFHpS |
5.38 |
449.0 |
80.0 |
86 |
-18 |
90 |
Qualifier |
Analyte |
| PFHpS |
5.38 |
449.0 |
99.0 |
52 |
-18 |
80 |
Qualifier |
Analyte |
| 13C4-PFOA |
5.39 |
417.0 |
372.0 |
17 |
-5 |
80 |
Quantifier |
NIS |
| PFOA |
5.39 |
413.0 |
369.0 |
20 |
-10 |
68 |
Quantifier |
Analyte |
| PFOA |
5.39 |
413.0 |
169.0 |
30 |
-10 |
80 |
Qualifier |
Analyte |
| 13C8-PFOA |
5.39 |
421.0 |
376.0 |
16 |
-4 |
84 |
Quantifier |
EIS |
| 13C4-PFOS |
5.83 |
503.0 |
80.0 |
105 |
-45 |
125 |
Quantifier |
NIS |
| 13C8-PFOS |
5.84 |
507.0 |
80.0 |
107 |
-22 |
140 |
Quantifier |
EIS |
| 13C8-PFOS |
5.84 |
507.0 |
99.0 |
50 |
-22 |
140 |
Qualifier |
EIS |
| PFOS |
5.84 |
499.0 |
80.0 |
100 |
-50 |
170 |
Quantifier |
Analyte |
| PFOS |
5.84 |
499.0 |
99.0 |
55 |
-50 |
170 |
Qualifier |
Analyte |
| 13C9-PFNA |
5.85 |
472.0 |
427.0 |
20 |
-10 |
75 |
Quantifier |
EIS |
| 13C5-PFNA |
5.85 |
468.0 |
423.0 |
18 |
-14 |
70 |
Quantifier |
NIS |
| PFNA |
5.85 |
463.0 |
419.0 |
20 |
-10 |
75 |
Quantifier |
Analyte |
| PFNA |
5.85 |
463.0 |
219.0 |
29 |
-10 |
90 |
Qualifier |
Analyte |
| 7:3FTCA |
6.03 |
441.0 |
317.0 |
35 |
-10 |
190 |
Quantifier |
Analyte |
| 7:3FTCA |
6.03 |
441.0 |
337.0 |
24 |
-10 |
190 |
Qualifier |
Analyte |
| 9Cl-PF3ONS |
6.05 |
533.0 |
351.0 |
35 |
-30 |
110 |
Quantifier |
Analyte |
| 9Cl-PFONS |
6.05 |
533.0 |
83.0 |
35 |
-30 |
119 |
Qualifier |
Analyte |
| PFNS |
6.23 |
549.0 |
80.0 |
80 |
-5 |
110 |
Quantifier |
Analyte |
| 13C6-PFDA |
6.25 |
519.0 |
474.0 |
17 |
0 |
88 |
Quantifier |
EIS |
| 13C2-PFDA |
6.25 |
515.0 |
470.0 |
16 |
-10 |
88 |
Quantifier |
NIS |
| PFDA |
6.25 |
513.0 |
469.0 |
21 |
-10 |
90 |
Quantifier |
Analyte |
| PFDA |
6.25 |
513.0 |
219.0 |
28 |
-10 |
96 |
Qualifier |
Analyte |
| 13C2-8:2FTS |
6.27 |
529.0 |
81.0 |
76 |
-40 |
115 |
Quantifier |
EIS |
| 13C2-8:2FTS |
6.27 |
529.0 |
509.0 |
35 |
-40 |
115 |
Qualifier |
EIS |
| 8:2FTS |
6.27 |
527.0 |
81.0 |
70 |
-3 |
100 |
Quantifier |
Analyte |
| 8:2FTS |
6.27 |
527.0 |
507.0 |
25 |
-30 |
90 |
Qualifier |
Analyte |
| d3-NMeFOSAA |
6.43 |
573.0 |
419.0 |
27 |
-25 |
105 |
Quantifier |
EIS |
| NMeFOSAA |
6.44 |
570.0 |
419.0 |
28 |
-20 |
100 |
Quantifier |
Analyte |
| NMeFOSAA |
6.44 |
570.0 |
483.0 |
19 |
-20 |
100 |
Qualifier |
Analyte |
| PFDS |
6.56 |
599.0 |
80.0 |
80 |
-3 |
130 |
Quantifier |
Analyte |
| 13C7-PFUnA |
6.58 |
570.0 |
525.0 |
21 |
-4 |
87 |
Quantifier |
EIS |
| PFUnA |
6.59 |
563.0 |
519.0 |
19 |
-10 |
96 |
Quantifier |
Analyte |
| PFUnA |
6.59 |
563.0 |
270.0 |
30 |
-10 |
85 |
Qualifier |
Analyte |
| d5-NEtFOSAA |
6.6 |
589.0 |
419.0 |
28 |
-20 |
112 |
Quantifier |
EIS |
| d5-NetFOSAA |
6.6 |
589.0 |
531.0 |
28 |
-20 |
105 |
Qualifier |
EIS |
| NEtFOSAA |
6.6 |
584.0 |
419.0 |
30 |
-20 |
100 |
Quantifier |
Analyte |
| NetFOSAA |
6.6 |
584.0 |
482.0 |
22 |
-20 |
99 |
Qualifier |
Analyte |
| 13C8-PFOSA |
6.7 |
506.0 |
78.0 |
35 |
-5 |
110 |
Quantifier |
EIS |
| PFOSA |
6.7 |
498.0 |
78.0 |
48 |
-3 |
110 |
Quantifier |
Analyte |
| 11Cl-PF3OUdS |
6.71 |
631.0 |
451.0 |
37 |
-25 |
170 |
Quantifier |
Analyte |
| 11Cl-PF3OUdS |
6.71 |
631.0 |
199.0 |
32 |
-40 |
148 |
Qualifier |
Analyte |
| 13C2-PFDoA |
6.87 |
615.0 |
570.0 |
17 |
-14 |
104 |
Quantifier |
EIS |
| PFDoA |
6.87 |
613.0 |
569.0 |
17 |
-10 |
104 |
Quantifier |
Analyte |
| PFDoA |
6.87 |
613.0 |
319.0 |
30 |
-10 |
100 |
Qualifier |
Analyte |
| PFTrDA |
7.12 |
663.0 |
619.0 |
18 |
-6 |
104 |
Quantifier |
Analyte |
| PFTrDA |
7.12 |
663.0 |
369.0 |
40 |
-6 |
125 |
Qualifier |
Analyte |
| d7-NMeFOSE |
7.31 |
623.0 |
59.0 |
37 |
-11 |
83 |
Quantifier |
EIS |
| d5-NEtFOSA |
7.32 |
515.0 |
169.0 |
35 |
-3 |
110 |
Quantifier |
EIS |
| NMeFOSA |
7.32 |
512.0 |
219.0 |
30 |
-3 |
120 |
Quantifier |
Analyte |
| NMeFOSA |
7.32 |
512.0 |
169.0 |
30 |
-3 |
120 |
Qualifier |
Analyte |
| NMeFOSE |
7.32 |
616.0 |
59.0 |
35 |
-1 |
105 |
Quantifier |
Analyte |
| 13C2-PFTeDA |
7.33 |
715.0 |
670.0 |
18 |
-7 |
140 |
Quantifier |
EIS |
| PFDoS |
7.33 |
669.0 |
80.0 |
45 |
-5 |
110 |
Quantifier |
Analyte |
| PFDoS |
7.33 |
669.0 |
119.0 |
45 |
-5 |
100 |
Qualifier |
Analyte |
| PFTeDA |
7.33 |
713.0 |
669.0 |
19 |
-4 |
120 |
Quantifier |
Analyte |
| d9-NEtFOSE |
7.5 |
639.0 |
59.0 |
40 |
-11 |
84 |
Quantifier |
EIS |
| NEtFOSE |
7.51 |
630.0 |
59.0 |
19 |
-1 |
109 |
Quantifier |
Analyte |
| d3-NMeFOSA |
7.52 |
531.0 |
169.0 |
38 |
-2 |
110 |
Quantifier |
EIS |
| NEtFOSA |
7.52 |
526.0 |
219.0 |
30 |
-1 |
120 |
Quantifier |
Analyte |
| NEtFOSA |
7.52 |
526.0 |
169.0 |
30 |
-1 |
120 |
Qualifier |
Analyte |
a. CE = Collision Cell Energy
b. EV = Entrance Voltage
c. CCL2 = Collision Cell Lens 2 voltage
Calibration Standards Preparation
A mixed secondary stock of the target analytes was created from the five stock solutions, which was then used to generate ten calibration standards. Each calibration standard received an equal volume of Extracted Internal Standards (EIS) and Non-Extracted Internal Standards (NIS).
Based on the individual compound’s initial concentration in the stock solution, alyte concentration in calibration standards varied from 1 to 500,000 ng/L. For UHPLC analysis, calibration standards were moved to low-volume polypropylene vials with caps.
A wide range of calibration standards was employed to establish method linearity and instrument limits of detection (LOD). However, a smaller range and number of calibrants with a higher minimum level can be used in general practice. The EPA approach simply requires a minimum of six calibration standards.
Method Blanks, Fortified Samples, and Field Samples
All laboratory method blanks and fortified samples were created in 250 mL polypropylene bottles. A constant volume of EIS was spiked to each sample to assess extraction effectiveness based on recoveries.
A mixed analyte stock solution was utilized to spike known quantities of the target analytes into the fortified samples at different amounts to test and confirm analyte recoveries. This approach also determined the method detection limits (DL) and minimum level of quantitation (ML).
The SPE sample preparation procedure outlined in Section 12.0 of EPA Method 1633 extracted and concentrated all method blanks and fortified samples. Slight changes were made as permitted by the procedure.
The stacked GCB/WAX cartridge replaced the loose carbon step, reducing sample preparation time and the risk of analyte loss. The final 5 mL extracts were spiked with a consistent quantity of NIS before being transferred to vials with caps for analysis by LC/MS-MS.
Method blanks were routinely tested to guarantee the appropriate reduction or absence of PFAS interferences. Field samples were gathered from three non-potable sources in the Baltimore Metropolitan region. All field samples were collected in 250 mL polyethylene bottles and processed using SPE.
Solid Phase Extraction and Sample Concentration
PromoChrom SPE-003, an automated SPE system, was used. Extractions were carried out in line with the method defined in section 12.0 of EPA Method 1633. Stacked Weak Anion Exchange/Graphitized Carbon Black SPE 6 mL tubes containing 200 mg/50 mg sorbent, respectively.
The SPE approach followed the procedures shown in Figure 1. Because the method is performance-based, users elected to employ stacked GCB/ WAX cartridges instead of carbon cleanup, reducing both sample preparation time and the possibility of recovery losses.
Sample
Preparation |
- 250 mL sample
- Add EIS directly
- Check pH 6.0-7.0
|
Extraction
Setup |
- Silanized glass wool packed to half height of SPE cartridge
|
Condition
SPE |
- 15 mL of 1% methanolic ammonium hydroxide
- 5 mL of 0.3M formic acid
|
Load
Sample |
|
| Rinse |
- 5 mL of reagent water (x2)
- 5 mL 1:1 0.1M formic acid:MeOH
- Dry under vacuum for 15 sec
|
| Elute |
- Rinse sample bottle with 5 mL 1% NH4OH in MeOH
|
| Filter |
- Install a nylon syringe filter on a 5 mL poly syringe
- Decant sample supernatant into syringe barrel
|
Internal
Std |
- Add NIS to collection tube
|
| Analysis |
- Transfer an aliquot into AS vial
|
Figure 1. Steps of Solid Phase Extraction Method. Image Credit: PerkinElmer
Results and Discussion
Remediation of PFAS Background Contamination
One of the most significant issues associated with PFAS trace analysis is contamination from chemicals, SPE apparatus, sample collection materials, volumetric ware, vials, the LC/MS/MS system, and the lab environment.
Many of these interferences can be attributed to the materials used in the manufacturing of volumetric ware, pipettes, syringes, tubing, and vials, as well as from PTFE components in the LC/MS/MS system.
To prevent or reduce these interferences from the LC/MS/MS system, a delay column was installed between the pump’s mobile phase mixer and the autosampler’s sample valve to trap and delay any PFAS chemicals released by the pump and mobile phase solvents.
This effectively separates the PFAS chromatographic peaks in the sample from the incoming PFAS contaminant peaks from the pump system. To prevent potential contamination, the LX50 autosampler's regular PTFE tubing was replaced with PEEK tubing.
All the materials used in this research work were checked for PFAS contamination before running tests by injecting blank samples. These experiments proved that all of the supplies utilized were free of PFAS contamination.
LC and MS/MS Methods
The QSight MS/MS MRM parameters were optimized for each analyte, EIS, and NIS using direct infusion experiments with a syringe pump.
Wherever possible, one precursor and product mass were identified, and the entrance voltage (EV), collision cell energy (CE), and collision cell lens 2 voltage (CCL2) were optimized for each compound in Simplicity 3Q. Table 4 shows the optimal MRM parameters.
Once the retention periods for each analyte were determined, a time-managed MRM MS/MS technique was utilized, with optimized time windows and dwell times to ensure at least ten scans across each analyte peak. The LC gradient method was adjusted to achieve good analyte separation, reduce run time, and improve peak symmetry.
A high-efficiency superficially porous particle (SPP) column was selected to give narrow peaks and short run times.
Figure 2 shows the total ion chromatogram (TIC). Figure 3 depicts the initial demonstration of the LC method capabilities, which created a baseline separation of branched vs. linear isomers for PFHxS, PFOS, PFOA, NMeFOSAA, and NEtFOSAA.
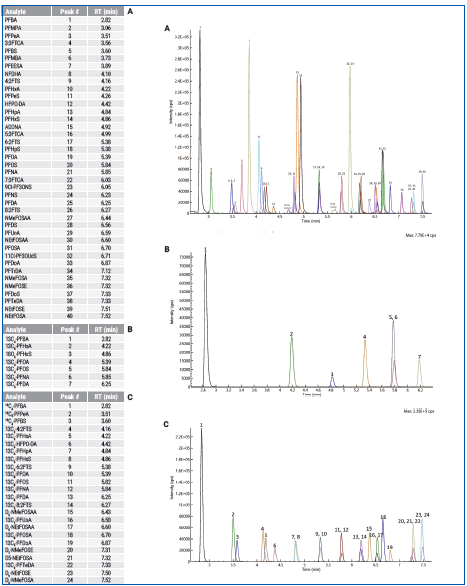
Figure 2. Total Ion Chromatogram of mid level calibration point containing (A) analytes, (B) non-extracted internal standards, and (C) extracted internal standards. Image Credit: PerkinElmer
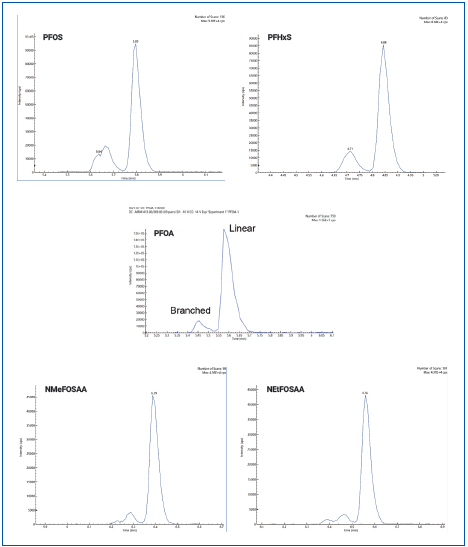
Figure 3. MRM chromatograms of PFHxS, PFOS, PFOA, NMeFOSAA, and NEtFOSAA showing the baseline separation of linear and branched chain isomers. Image Credit: PerkinElmer
Table 5. Instrument and Method Calibration Ranges and Linearity (R2) for eight-point calibration curves of all EPA Method 1633 analytes. Source: PerkinElmer
| Compound |
Instrument Calibration Range (ng/L)a |
R2b |
| PFBA |
4-100,000 |
0.99988 |
| PFMPA |
2-50000 |
0.99992 |
| PFPeA |
2-50000 |
0.99997 |
| 3:3FTCA |
3-70000 |
0.99720 |
| PFBS |
1-25000 |
0.99989 |
| PFMBA |
2-50000 |
0.99984 |
| PFEESA |
2-50000 |
0.99988 |
| NFDHA |
2-50000 |
0.99965 |
| 4:2FTS |
4-100000 |
0.99953 |
| PFHxA |
1-25000 |
0.99983 |
| PFPeS |
1-25000 |
0.99989 |
| HFPO-DA |
2-50000 |
0.99909 |
| PFHpA |
1-25000 |
0.99980 |
| PFHxS |
1-25000 |
0.99951 |
| ADONA |
2-50000 |
0.99958 |
| 5:3FTCA |
15-350000 |
0.99972 |
| 6:2FTS |
4-100000 |
0.99900 |
| PFHpS |
1-25000 |
0.99968 |
| PFOA |
1-25000 |
0.99989 |
| PFOS |
1-25000 |
0.99984 |
| PFOSA |
1-25000 |
0.99915 |
| PFNA |
1-25000 |
0.99936 |
| 7:3FTS |
15-350000 |
0.99949 |
| 9Cl-PF3ONS |
2-50000 |
0.99990 |
| PFNS |
1-25000 |
0.99968 |
| PFDA |
1-25000 |
0.99947 |
| 8:2FTS |
4-100000 |
0.99945 |
| NMeFOSAA |
1-25000 |
0.99969 |
| PFDS |
1-25000 |
0.99952 |
| PFUnA |
1-25000 |
0.99984 |
| NEtFOSAA |
1-25000 |
0.99941 |
| 11Cl-PF3OUdS |
2-50000 |
0.99901 |
| PFDoA |
1-25000 |
0.99915 |
| PFTrDA |
1-25000 |
0.99925 |
| NMeFOSA |
1-25000 |
0.99966 |
| NMeFOSE |
10-250000 |
0.99984 |
| PFDoS |
1-25000 |
0.99914 |
| PFTeDA |
1-25000 |
0.99928 |
| NEtFOSE |
10-250000 |
0.99966 |
| NEtFOSA |
1-25000 |
0.99934 |
a. Instrument calibration range is the actual concentration range of calibration
standards used to determine calibration curves.
b. R2 values are the average of triplicate calibration curves.
Table 6. Instrument limits of detection (LOD) and limits of quantitation (LOQ) for all target analytes in EPA Method 1633. Source: PerkinElmer
| Analyte |
Instrument (ng/L)a |
| LOD |
LOQ |
| PFBA |
10 |
30 |
| PFMPA |
1 |
4 |
| PFPeA |
5 |
15 |
| 3:3FTCA |
50 |
160 |
| PFBS |
1 |
4 |
| PFMBA |
3 |
9 |
| PFEESA |
2 |
5 |
| NFDHA |
1 |
4 |
| 4:2FTS |
1 |
4 |
| PFHxA |
4 |
12 |
| PFPeS |
1 |
4 |
| HFPO-DA |
20 |
60 |
| PFHpA |
0.5 |
2 |
| PFHxS |
1 |
4 |
| ADONA |
0.8 |
3 |
| 5:3FTCA |
0.3 |
1 |
| 6:2FTS |
4.2 |
14 |
| PFHpS |
1 |
3 |
| PFOA |
2 |
5 |
| PFOS |
0.5 |
2 |
| PFNA |
6 |
20 |
| 7:3FTCA |
5 |
17 |
| 9Cl-PF3ONS |
10 |
34 |
| NMeFOSAA |
6 |
21 |
| PFNS |
2 |
6 |
| PFDA |
20 |
65 |
| 8:2FTS |
4 |
13 |
| NEtFOSAA |
4 |
12 |
| PFDS |
1 |
4 |
| PFUnA |
10 |
45 |
| 11Cl-PF3OUdS |
4 |
14 |
| PFOSA |
0.5 |
2 |
| PFDoA |
20 |
67 |
| PFTrDA |
9 |
29 |
| PFDoS |
30 |
87 |
| PFTeDA |
4 |
14 |
| NMeFOSA |
7 |
24 |
| NMeFOSE |
20 |
53 |
| NEtFOSE |
5 |
16 |
| NEtFOSA |
8 |
25 |
a. Instrument LOD/LOQ was determined using the signal-to-noise ratio (S/N) of the peak from the lowest detectable calibration standard and extrapolating to the concentration at which the S/N = 3 or 10 for LOD or LOQ, respectively. This is an estimate to demonstrate expected LOD/LOQ and can vary from lab to lab.
Linearity, Instrument Limits of Quantitation (LOQ), and Instrument Limits of Detection (LOD)
Calibration curves were utilized to determine linearity, instrument limits of detection (LOD), and limits of quantitation (LOQ) for all PFAS targets and surrogates.
Three replicates were used to create ten-point calibration curves in the concentration range of ~1 – 500,000 ng/L, using non-weighted linear regression with the intercept set to zero.
Table 5 shows that all analytes had correlation coefficient values (R2) of more than 0.99, indicating excellent linearity over the studied range of concentrations.
The instrument limits of detection (LOD) and quantitation (LOQ) for each target analyte were established at the lowest detectable standard on the calibration curve (ng/L), which was then extrapolated to yield a signal-to-noise ratio (S/N) of 3 for LOD and 10 for LOQ.
Table 6 summarizes the instrument and method limits of detection (LODs) and limits of quantification (LOQs).
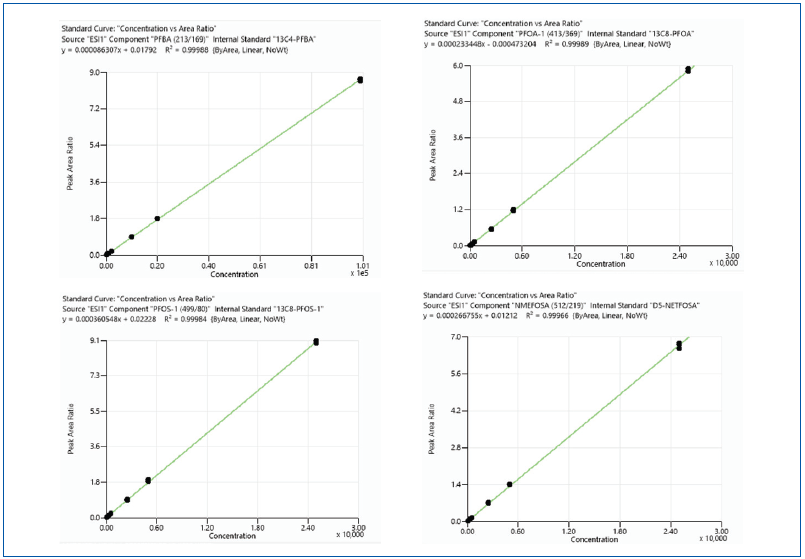
Figure 4. Triplicate injection calibration curves for representative analytes PFBA, PFOA, PFOS, NMeFOSA. Image Credit: PerkinElmer
Determination of Method DLs, MRLs
The method detection limits (DL) and minimum quantitation (ML) levels were calculated using EPA Method 1633. Replicate reagent water samples were spiked at three varying levels and used in computations. Each fortified sample received a constant volume of EIS.
Each of these fortified samples was processed using the full sample preparation approach. Aliquots of each sample were then moved to polypropylene vials and examined using an LC/MS/MS system to measure analyte and EIS recoveries.
Table 7 shows that the recoveries for all analytes at all fortification levels ranged between 94 and 106%. The recovery RSDs were less than 10% across all fortification levels. All recoveries were within the limits specified by the EPA method for the extracted internal standards.
The method DLs and MLs were computed and validated using the statistical analysis approaches outlined in EPA Method 1633. Table 8 presents the determinations of DLs and MLs.
Table 7. PFAS analyte recovery data for fortified of reagent water spiked at two different levels. Five replicate samples were extracted at each fortification level. Source: PerkinElmer
| |
Low Fortification Level (n=5) |
High Fortification Level (n=5) |
Fortified
Conc.
(ng/L) |
%
Average
Recovery |
%RSD |
Fortified
Conc.
(ng/L) |
%
Average
Recovery |
%RSD |
| PFBA |
16 |
100 |
4 |
64 |
106 |
2 |
| PFMPA |
8 |
106 |
2 |
32 |
99 |
6 |
| PFPeA |
8 |
97 |
3 |
32 |
99 |
5 |
| 3:3FTCA |
16 |
103 |
6 |
64 |
99 |
5 |
| PFBS |
4 |
101 |
7 |
16 |
98 |
5 |
| PFMBA |
8 |
94 |
5 |
32 |
99 |
3 |
| PFEESA |
8 |
96 |
4 |
32 |
98 |
5 |
| NFDHA |
8 |
99 |
6 |
32 |
101 |
3 |
| 4:2FTS |
16 |
96 |
2 |
64 |
94 |
3 |
| PFHxA |
4 |
99 |
6 |
16 |
97 |
4 |
| PFPeS |
4 |
99 |
5 |
16 |
98 |
4 |
| HFPO-DA |
8 |
97 |
3 |
32 |
98 |
4 |
| PFHxS |
4 |
98 |
4 |
16 |
99 |
2 |
| PFHpA |
4 |
101 |
3 |
16 |
103 |
3 |
| ADONA |
8 |
101 |
3 |
32 |
100 |
2 |
| 5:3FTCA |
80 |
98 |
4 |
320 |
95 |
2 |
| 6:2FTS |
16 |
99 |
3 |
64 |
98 |
4 |
| PFOA |
4 |
101 |
2 |
16 |
100 |
2 |
| PFHpS |
4 |
103 |
5 |
16 |
103 |
2 |
| PFOS |
4 |
98 |
4 |
16 |
96 |
5 |
| PFNA |
4 |
102 |
1 |
16 |
101 |
3 |
| 7:3FTCA |
80 |
104 |
2 |
320 |
104 |
3 |
| 9Cl-PF3ONs |
8 |
98 |
4 |
32 |
99 |
4 |
| NMeFOSAA |
4 |
97 |
3 |
16 |
95 |
3 |
| PFNS |
4 |
98 |
4 |
16 |
101 |
3 |
| PFDA |
4 |
100 |
4 |
16 |
99 |
3 |
| 8:2FTS |
16 |
101 |
3 |
64 |
99 |
4 |
| NEtFOSAA |
4 |
99 |
2 |
16 |
98 |
4 |
| PFDS |
4 |
100 |
4 |
16 |
101 |
2 |
| PFUnA |
4 |
95 |
3 |
16 |
102 |
3 |
| 11Cl-PF3OUdS |
8 |
99 |
3 |
32 |
98 |
2 |
| PFOSA |
4 |
95 |
6 |
16 |
99 |
3 |
| PFDoA |
4 |
99 |
5 |
16 |
100 |
5 |
| PFTrDA |
4 |
102 |
6 |
16 |
99 |
4 |
| PFDoS |
4 |
97 |
5 |
16 |
100 |
5 |
| PFTeDA |
4 |
99 |
5 |
16 |
97 |
4 |
| NMeFOSA |
4 |
99 |
4 |
16 |
100 |
2 |
| NMeFOSE |
40 |
98 |
3 |
160 |
96 |
4 |
| NEtFOSE |
40 |
99 |
5 |
160 |
98 |
4 |
| NEtFOSA |
4 |
100 |
5 |
16 |
100 |
3 |
Table 8. Method detection limits (MDL) and minimum level of quantitation (ML) on the QSight 210 LC-MS/MS system. Experimental values are compared to reference values reported in EPA Method 1633. Source: PerkinElmer
| Analyte |
QSight 210 |
EPA Method 1633c |
| MDLa (ng/L) |
MLb (ng/L) |
MDL (ng/L) |
ML (ng/L) |
| PFBA |
1.33 |
5.0 |
0.79 |
2.0 |
| PFMPA |
0.43 |
2.0 |
1.46 |
5.0 |
| PFPeA |
1.21 |
5.0 |
0.54 |
2.0 |
| 3:3FTCA |
3.04 |
10.0 |
2.47 |
10.0 |
| PFBS |
1.30 |
5.0 |
0.37 |
2.0 |
| PFMBA |
1.16 |
5.0 |
1.41 |
5.0 |
| PFEESA |
0.35 |
2.0 |
1.17 |
5.0 |
| NFDHA |
0.41 |
2.0 |
0.75 |
2.0 |
| 4:2FTS |
1.16 |
5.0 |
1.68 |
5.0 |
| PFHxA |
0.96 |
5.0 |
0.46 |
2.0 |
| PFPeS |
0.63 |
2.0 |
0.5 |
2.0 |
| HFPO-DA |
0.89 |
5.0 |
0.51 |
2.0 |
| PFHxS |
0.56 |
2.0 |
0.54 |
2.0 |
| PFHpA |
0.58 |
2.0 |
0.37 |
2.0 |
| ADONA |
0.71 |
2.0 |
0.50 |
2.0 |
| 5:3FTCA |
0.25 |
2.0 |
9.59 |
20.0 |
| 6:2FTS |
1.24 |
5.0 |
2.45 |
10.0 |
| PFOA |
1.10 |
5.0 |
0.54 |
2.0 |
| PFHpS |
1.12 |
5.0 |
0.50 |
2.0 |
| PFOS |
0.71 |
2.0 |
0.63 |
2.0 |
| PFNA |
1.04 |
2.0 |
0.45 |
2.0 |
| 7:3FTCA |
1.04 |
2.0 |
8.71 |
20.0 |
| 9Cl-PF3ONS |
3.51 |
10.0 |
1.38 |
5.0 |
| NMeFOSAA |
0.31 |
2.0 |
0.68 |
2.0 |
| PFNS |
0.30 |
2.0 |
0.47 |
2.0 |
| PFDA |
2.94 |
10.0 |
0.52 |
2.0 |
| 8:2FTS |
0.74 |
2.0 |
2.5 |
10.0 |
| NEtFOSAA |
0.77 |
2.0 |
0.59 |
2.0 |
| PFDS |
0.61 |
2.0 |
0.60 |
2.0 |
| PFUnA |
0.56 |
2.0 |
0.45 |
2.0 |
| 11Cl-PF3OUdS |
0.69 |
2.0 |
1.67 |
5.0 |
| PFOSA |
0.79 |
5.0 |
0.32 |
2.0 |
| PFDoA |
1.24 |
5.0 |
0.40 |
2.0 |
| PFTrDA |
0.51 |
2.0 |
0.46 |
2.0 |
| PFDoS |
2.86 |
10.0 |
0.6 |
2.0 |
| PFTeDA |
0.65 |
2.0 |
0.49 |
2.0 |
| NMeFOSA |
0.51 |
2.0 |
0.43 |
2.0 |
| NMeFOSE |
0.46 |
2.0 |
3.81 |
10.0 |
| NEtFOSE |
0.62 |
2.0 |
4.84 |
20.0 |
| NEtFOSA |
0.48 |
2.0 |
0.45 |
2.0 |
a. Experimental DL was determined from seven LFB replicates fortified at ~4-16 ng/L measured over three days and calculated according to procedure 40 CFR Part 136, Appendix B.
b. Experimental MLs were calculated by multiplying the MDL by 3.18 and rounding to the nearest, 1, 2 or 5 x 10n as specified in section 9.2.2 of 1633.
c. Values taken directly from Table 8 of 1633 Draft 4.
Field Sample Analysis
Field samples were gathered from three distinct locations in the Baltimore Metropolitan region and labeled S1, S2, and S3. Three samples were taken from each location. Before extraction, all samples were spiked with a consistent amount.
All samples collected at a single location were extracted, evaporated, and reconstituted using SPE technology. The reconstituted samples were subsequently spiked with NIS, and an aliquot was moved to a polypropylene vial for LC/MS/MS testing.
Table 9 summarizes the results from all samples. At least one PFAS compound was identified in each sample. The first sample contained PFOS, while the second sample had PFBA, PFBS, PFHpA, and PFOS. PFBA and PFHpA were detected in sample 3.
All other analytes were either undetectable or below the MLs, as indicated by <ML in the table.
Conclusion
This article describes the validation of an LC/MS/MS technique for determining PFAS analytes and isotopically labeled standards listed in the US EPA Method 1633. This technique uses the PerkinElmer LX50 UHPLC System in conjunction with the PerkinElmer QSight 210 Triple Quadrupole Mass Spectrometer.
All PFAS analytes and isotopic standards demonstrated excellent linearity, with R2 values of ≥ 0.99. The instrument LODs and LOQs confirm that the QSight 210 LC/MS/MS System possesses the necessary sensitivity to quantify the PFAS analytes.
To eliminate and minimize background PFAS pollutants, instrument modifications and the insertion of a delay column are required, and the effectiveness of these modifications has been verified using blank analysis.
An enhanced chromatographic technique previously developed for EPA Method 537.1 to reduce LC/MS/MS runtimes has been demonstrated to work for 533 analysis and is now being used for 1633 analysis.14,15
The QSight 210 LC/MS/MS System was used to optimize MRM experiments for all analytes and isotopically labeled standards, including quantifier and qualifier MRMs where feasible.
A time-managed MRM mass spectrometer approach has also been optimized to enhance dwell time for increased sensitivity while retaining more than ten data points per chromatographic peak.
Recoveries for all analytes range between 94 and 106%, while recoveries for extracted internal standards all fall within the values defined by method 1633. The SPE extraction in the current study was performed using an automated SPE system. Non-potable water samples from three separate locations were collected and examined.
Overall, this validation study demonstrates that the LX50 UHPLC system combined with the QSight 210 tandem quadrupole mass spectrometer is an effective analytical instrument for the application of EPA Method 1633, with sufficient sensitivity to assess all analytes.
In addition, a singular method, which used the same columns and mobile phases, was validated for analyzing all analytes from EPA Methods 1633, 533, and 537.1 without physically altering the system or consumables other than using a different SPE cartridge and preparation technique as specified by each methodology.
This will significantly enhance throughput in labs that need to analyze drinking water using either EPA method.
Table 9. Average concentration of PFAS from each sampling location. Source: PerkinElmer
| Analyte |
Average Concentration |
| S1 |
S2 |
S3 |
| PFBA |
<ML |
12±2 |
13±1 |
| PFMPA |
<ML |
<ML |
<ML |
| PFPeA |
<ML |
<ML |
<ML |
| 3:3FTCA |
<ML |
<ML |
<ML |
| PFBS |
<ML |
1.5±0.5 |
<ML |
| PFMBA |
<ML |
<ML |
<ML |
| PFEESA |
<ML |
<ML |
<ML |
| NFDHA |
<ML |
<ML |
<ML |
| 4:2FTS |
<ML |
<ML |
<ML |
| PFHxA |
<ML |
<ML |
<ML |
| PFPeS |
<ML |
<ML |
<ML |
| HFPO-DA |
<ML |
<ML |
<ML |
| PFHxS |
<ML |
<ML |
<ML |
| PFHpA |
<ML |
0.7±0.1 |
0.62±0.02 |
| ADONA |
<ML |
<ML |
<ML |
| 5:3FTCA |
<ML |
<ML |
<ML |
| 6:2FTS |
<ML |
<ML |
<ML |
| PFOA |
<ML |
<ML |
<ML |
| PFHpS |
<ML |
<ML |
<ML |
| PFOS |
2.0±0.3 |
8±1 |
<ML |
| 7:3FTCA |
<ML |
<ML |
<ML |
| 9Cl-PF3ONS |
<ML |
<ML |
<ML |
| NMeFOSAA |
<ML |
<ML |
<ML |
| PFNS |
<ML |
<ML |
<ML |
| PFDA |
<ML |
<ML |
<ML |
| 8:2FTS |
<ML |
<ML |
<ML |
| NEtFOSAA |
<ML |
<ML |
<ML |
| PFDS |
<ML |
<ML |
<ML |
| PFUnA |
<ML |
<ML |
<ML |
| 11Cl-PF3OUdS |
<ML |
<ML |
<ML |
| PFOSA |
<ML |
<ML |
<ML |
| PFDoA |
<ML |
<ML |
<ML |
| PFTrDA |
<ML |
<ML |
<ML |
| PFDoS |
<ML |
<ML |
<ML |
| PFTeDA |
<ML |
<ML |
<ML |
| NMeFOSA |
<ML |
<ML |
<ML |
| NMeFOSE |
<ML |
<ML |
<ML |
| NEtFOSE |
<ML |
<ML |
<ML |
| NEtFOSA |
<ML |
<ML |
<ML |
References and Further Reading
- Kissa, E., Fluorinated Surfactants and Repellents. Second Edition ed. Surfactant Science Series, ed. A. Hubbard. 2001, New York, NY: Marcel Dekker.
- Lau, Christopher, et. al. “Perfluoroalkyl Acids: A Review of Monitoring and Toxicological Findings.” Toxicological Sciences, 99, 2, 2007, pp 366-394. http://doi.org/10.1093/toxsci/kfm128
- https://www.epa.gov/pfas/basic-information-pfas
- Wang, Z. et al. “An Overview of the Uses of PFAS.” https://doi.org/10.1039/D0EM00291G
- Boiteux, V.; Bach, C.; Sagres, V.; Hemard, J.; Colin, A.; Rosin, C.; Munoz, J.-F.; Dauchy, X. Analysis of 29 Per- and Polyfluorinated Compounds in Water, Sediment, Soil and Sludge by Liquid Chromatography–Tandem Mass Spectrometry. Int. J. Environ. Anal. Chem. 2016, 96 (8), pp 705–728. https://www.tandfonline.com/doi/full/10.1080/03067319.2016.1196683
- Banzhaf, S.; Filipovic, M.; Lewis, J.; Sparrenbom, C.J.; Barthel, R. “A Review of Contamination of Surface-, Ground-, and Drinking Water in Sweden by Perfluoroalkyl and Polyfluoroalkyl Substances (PFASs).” Ambio. Springer Netherlands April 1, 2017, pp 335-346. https://doi.org/10.1007/s13280-016-0848-8
- Buck, R. C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousnis, I.T.; Voogt, P. De’ Jensen, A.A.; Kaanan, K.; Mabury, S.A.; van Leeuwen, S.P.J. “Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins.” Integr. Environ. Assess. Manag. 2011, 7(4), pp 513-541. https://setac.onlinelibrary.wiley.com/doi/10.1002/ieam.258#:~:text=(2006)%2C%20the%20presence%20of,of%20larger%20functional%20derivatives%20and
- Parry, E.; Anumol, T. “Quantitative Analysis of PFAS in Drinking Water Using Liquid Chromatography Tandem Mass Spectrometry.” The Column 2019, 15 (5), pp 9–15.
- Glaser, J.A.; Foerst, D.L.; McKee, G.D.; Quave, S.A.; Budde, W.L. “Trace Analyses for Wastewaters.” Environ. Sci. Technol. 1981, 15, pp 1426-1435.
- Maya E. Morales-McDevitt, Jitka Becanova, Arlene Blum, Thomas A. Bruton, Simon Vojta, Melissa Woodward, and Rainer Lohmann. Environmental Science & Technology Letters 2021 8 (10), 897-902. DOI: 10.1021/acs.estlett.1c00481
- Shoemaker, J.A.; Tettenhorst, D.R. “EPA Method 537.1: Determination of Selected Per- and Polyflourinated Alkyl Substances in Drinking Water by Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry (LC/ MS/MS) Version 2.0” EPA Document #: EPA/600/R-20/006 March 2020.
- Rosenblum, L., Wendelken, S.C. “EPA Method 533: Determination of Per- and Polyfluoroalkyl Substances in Drinking Water by Isotope Dilution Anion Exchange Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry” EPA Document #: 815-B-19-020 December 2019.
- “Draft Method 4 1633 Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous, Solid, and Biosolids, and Tissue Samples by LC-MS/MS” EPA Document #: EPA 821-D-23-001 July 2023.
- Weisenseel, J., Costanzo, M. “Analysis of Perfluoroalkyl and Polyfluoroalkyl Substances in Drinking Water: Validation Studies of EPA Method 537.1 Using the QSight 220 UHPLC/ MS/MS” Application Note.
- Belunis, A., LaCourse, W., Costanzo, M. “Improved Throughput for the Analysis of Perfluoroalkyl and Polyfluoroalkyl Substances in Drinking Water by EPA Method 533” Application Note.

This information has been sourced, reviewed and adapted from materials provided by PerkinElmer.
For more information on this source, please visit PerkinElmer.