The article provides a detailed overview of the process used to determine leachables and extractables from plastics. This process is mostly preferred for plastics employed in food contact applications and in biomedical devices. The following topics are described:
- Definitions
- The purpose of E&L testing
- Why is E&L testing important
- Regulatory Environment
- What are extractables and leachables?
- How is a E&L study conducted?
- Sample selection
- Sample preparation
- Extraction conditions
- Identification of E&Ls (Qualitative Analysis)
- Determination of E&L concentration (Quantitative Analysis)
- Acceptable levels for E&Ls
- Quality control in an E&L study
Definitions
E&L Study
Extractables and Leachables study
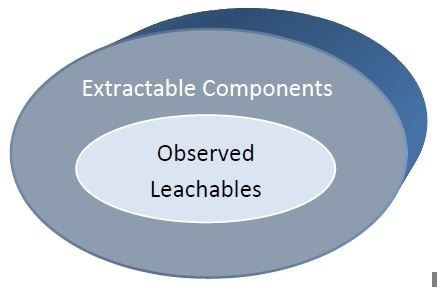
Leachable
"substances that can be released from a medical device or material during clinical use1”
Extractable
"substances that can be released from a medical device or material using extraction solvents and/or extraction conditions that are expected to be at least as aggressive as the conditions of clinical use1”
The Purpose of E&L Testing
Almost 20% of the US adult population has an implanted medical device. Plastics are extensively used in medical devices, covering almost all types of devices. Different types of plastics are used in these devices and they include polymers such as polyurethanes, polycarbonate, silicones, polyethylene, polyvinylchloride (PVC) etc. Beyond the base polymer, plastics also contain additional components.
These components contain a wide range of plastic additives that are used to enhance polymer properties and also byproducts and impurities that remain after polymer synthesis.
Residual monomer and oligomers (polymer chains <2000 Mw) are present in almost all polymers. When these small molecules are released from the plastic matrix, they can impart increased toxicity to the material. Identification of toxic small molecules present in the plastic and measurement of their quantity is made through extractables and leachables (E&L) testing. The data obtained is then used along with toxicology data to assess health and safety concerns.
Why is E&L Testing Important?
The significance of E&L testing has been emphasized due to several highly publicized incidents. Earlier in 2009, a large product recall was carried out for Tylenol arthritis pain caplets after complaints related to a moldy smell. The problem was ultimately traced to the leaching of 2,4,6-tribromoanisole, a wood preservative which had transferred from wooden pallets to plastic packaging. 10 Other instances included increasing concern about components such as phthalate plasticizers that are generally found in PVC and Bisphenol A (BPA) from polycarbonate materials used in products for children. It has been shown that phthalate plasticizers can disrupt the endocrine system. In relation to these chemicals, the FDA has issued guidance and has limited the use of these chemicals for certain applications. 2, 3 Many other extractable components are also being investigated and some have been found to have deadly interactions with drugs.4
Regulatory Environment
The FDA has provided guidance in response to the hazards posed by E&L components. The guidance specifies that E&L testing should be performed as a part of 510(k) submissions for medical devices. 5, 6, 7 Besides this guidance, the "FDA Food Safety Modernization Act" also includes specific reference to E&L components, which states that, "A drug or device shall be deemed to be adulterated... if its container is composed, in whole or in part, of any poisonous or deleterious substance which may render the contents injurious to health." 8 This definition thus covers any small molecule component which may be released from the material and which has harmful effects (extractables). Additionally, the Product Quality Research Institute (PQRI) and other bodies have issued industry guidance documents, providing recommended maximum daily dosage levels for leachable components.
What are Extractables and Leachables?
So what exactly are leachables and extractables chemically? Leachables and extractables are the tiny molecules that are present in a polymer system including surfactants, antioxidants, plasticizers, slip agents, acid scavengers, lubricants, crosslinking agents, residual monomers and oligomers.
It is common to find tens or even hundreds of individual tiny molecules species which can be extracted from a given polymer system based on the extraction conditions. Most of these compounds may not be leachables based on their polarity as well as the device’s use conditions.
 Please click here if you would like more information on the product in this article or a quote
Please click here if you would like more information on the product in this article or a quote
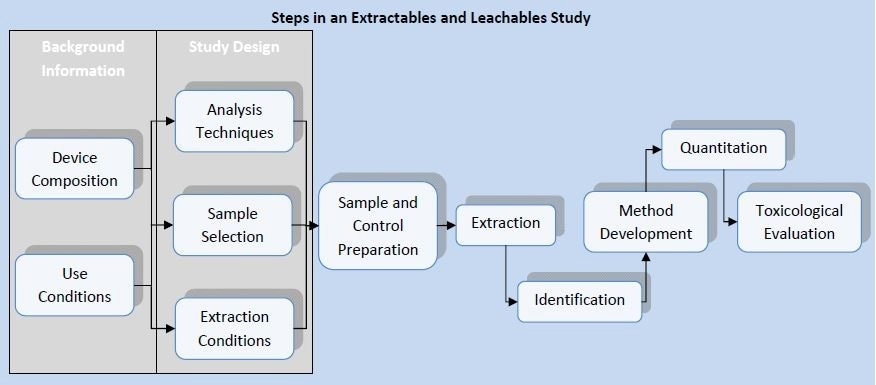
How is an E&L Study Conducted?
ISO 10993-12 provides guidance for the proper procedure for executing leachables and extractables studies. ISO 10993-18 provides more information about the proper selection of analytical methods for chemical characterization. This set of documents defines the topics which should be taken into account when conducting an E&L study. The key steps include:
- Sample selection
- Sample preparation
- Extraction
- Qualitative analysis
- Quantitative analysis
Sample Selection
When it comes to sample selection, it is important to ensure that the specimen is representative of the end product since it will be applied to the food contact application or patient. E&Ls can be picked up or created during the manufacturing process. To accurately simulate the risk to the patient, samples containing all the same E&Ls as those found in the actual device should be utilized. For this reason it is preferred that the actual medical device be utilized for testing whenever possible. If device sterilization is needed, then the E&L study should be carried out after sterilization. When using the actual device is not practical, composite samples can also be prepared but special care should be taken to make sure that the sample is representative of the end product. Extraction of separate sample components can also be helpful for determining the source of individual E&Ls.
Sample Preparation
Generally, less sample preparation is needed when a medical device is tested in its entirety. During the analysis of the entire device, the device is submerged into the extraction solvent or the device is filled as appropriate. Sample preparation in this case involves creating appropriate enclosures to block non-wetted regions of the device.
If it is impractical to analyze the entire device, then representative parts of the device can be utilized. This often involves cutting or machining parts of each of the materials that are used to make up the device. Collecting the materials from a finished device is generally preferred as this will ensure that the materials were subjected to the same manufacturing conditions. It must be ensured that each material present in the medical device is included in proportion to its quantity in the device. Importance should be given to those components which are known to show a biological response. It is advised that plastics be cut into portions with dimensions of 10 mm x 50 mm or smaller in order to enhance extraction efficiency.
Extraction Conditions
Before selecting the optimum extraction conditions, it is important to have a good understanding of the conditions in which the device will be used. The guiding principle when choosing the optimum extraction conditions is that the extraction should provide a suitable exaggeration of the predicted conditions of product use. This gives a margin of safety when evaluating the leachables which can be predicted to come from a device. Typically, three different types of extractions are applied and are described in ISO-10993-12:
Simulated-use Extraction
An extraction conducted using a method that simulates the expected use conditions.
Exaggerated Extraction
An extraction which uses conditions which are expected to cause a greater amount of extractable material to be released than using the simulated use extraction.
Exhaustive Extraction
An extraction which is repeated until the total amount of extractables is less than 10% of the amount obtained during the initial extraction.
In majority of cases, a simulated extraction and an exhaustive or exaggerated extraction will be carried out. The simulated use extracts are examined to find out what can practically be expected to leach from the sample under actual use conditions. The exaggerated or exhaustive extract is used to estimate the highest amount of extractable material which can extract from the sample under worst case conditions. In exhaustive extractions, multiple extractions have to be carried out on a single sample. This can be achieved using Soxhlet extraction or a conventional extraction approach and applying multiple cycles. In both cases, extraction completeness has to be verified.
A range of factors such as solvent type, temperature, time, surface-area-to-volume ratio and agitation conditions affect the extraction process. The optimum conditions are a function of the nature of the sample, the type of extraction which is preferred, and the analytical methods which will be applied to find out the chemistry of the extracted materials. Another aspect that needs to be considered is the intended use conditions for the device.
Extractions should be carried out in both a non-polar and polar solvent. Typical non-polar solvents include hexane and chloroform, and typical polar solvents include saline or water. Generally, the solvent should be chosen such that the amount of extractables is maximized without dissolving the polymer itself. Also, solvent volatility should be considered as it is usually preferred to concentrate the extracted components to increase method sensitivity. It is important that the extraction solvents are compatible with the analytical methods that are being applied to detect the extracted components. It is equally important that the extraction conditions do not cause changes in the sample chemistry. Throughout the extraction process, the sample solution should be agitated.
Either a mass basis or the surface area of the component being extracted can be used to determine the amount of extraction solvent utilized. As a rule, the surface area approach is favored and a surface area of volume ratio of 3 cm2/ml is applied for majority of samples. Higher volumes may prove suitable for high surface area materials such as films and sheets.
The extraction temperature relies on the intended use conditions and is chosen to provide a proper exaggeration of the expected use conditions. One of four temperatures are usually applied.
- (37 ± 1) °C for (72 ± 2) hours
- (50 ± 2) °C for (72 ± 2) hours
- (70 ± 2) °C for (24 ± 2) hours
- (121 ± 2) °C for (1 ± 0,1) hours
It is advised to analyze the prepared extract as soon as is reasonable. If the extract is kept longer than 24 hours, then it is important to consider the homogeneity and stability of the extract. Before analysis, concentration of the extract should be done to maximize method sensitivity. This concentration step should be carefully considered because analyte loss can occur for volatile or unstable components during the time of concentration.
Identification of E&Ls (Qualitative Analysis)
After obtaining an extract, appropriate analytical techniques should be used to identify the chemistry of the components extracted. It would be ideal if this process is initiated by reviewing the materials used to manufacture the device, because the expected starting materials and additives which may be present can then be determined. This would make it possible to perform targeted analyzes as well as confirmation of the applicability of analytical methods and methodologies.
Types of Unknowns
The methodologies and the analytical techniques used are crucial to the success of an E&L study. If the selected test methods lack adequate scope, then components that were successfully extracted will not be detected. Analytical techniques have certain limitations, which are not known to many device manufacturers and they think that a single method can sufficiently identify unknowns in an extract.
Unknowns fall into a number of broad classes such as volatiles, non-volatile and semi-volatile components. Also, extracted components can be organic, such as additives and monomers, or inorganic, such as metals or salts. Generally, the following analytical techniques are used to determine the different classes of unknowns:
Organic Unknowns
Volatile
Gas Chromatography Mass Spectroscopy (GCMS), Dynamic Headspace GCMS (D-HMS), Headspace GCMS (HGCMS), Desorption Mass Spectroscopy (DMS)
Non-Volatile
Nuclear Magnetic Resonance (NMR), Pyrolysis Mass Spectroscopy (PYMS), Gel Permeation Chromatography (GPC), Fourier Transform Infrared Spectroscopy (FTIR)
Semi-Volatile
Liquid Chromatography Mass Spectroscopy (LCMS), Gas Chromatography Mass Spectroscopy (GCMS), Desorption Mass Spectroscopy (DMS)
Inorganic Unknowns
Metals
X-ray Fluorescence (XRF), Inductively Coupled Mass Spectroscopy (ICP-MS)
Salts
Inductively Coupled Mass Spectroscopy (ICP-MS), Ion Exchange Chromatography (IEC)
There are other methods that can also be used for targeted analyzes of expected components.
The use of appropriate analytical techniques is very important, but this is just a starting point for a successful analysis. Also, proper identification of unknowns is based on the interpretation of data acquired to corroborate the chemistry of each unknown. In case a compound is not commercially available, then a definitive identification cannot be obtained without a great deal of analytical effort. In fact, the accuracy of unknown identification is largely dependent on the analyst’s skill and involves a combination of factors such as the use of spectral and compound database searches, manual data interpretation and an understanding of how different types of methods can be applied to corroborate one another. Also, the quality of the analytical data can differ considerably based on the exact instrument type and suitability, the analysis conditions, and the analyst’s knowledge of the chemistry of the materials (background information). All these factors place a great deal of importance on the selection of the laboratory utilized to perform the analyzes.
Determination of E&L Concentration (Quantitative Analysis)
The potential hazards associated with leachables can be assessed by determining the concentration released from the plastic. However, this is quite difficult because of the wide range of leachables and extractables as well as the need for proper identification. If the components are not commercially available, identification beyond acquiring a molecular formula may not be possible. This is particularly true in cases where the material is a reaction byproduct or degradant from the polymer synthesis. Considering these reasons, a number of different strategies should be applied to quantitate the components identified in an E&L study. Based on experience, two main methods are applied for quantitation — formal quantitation using standards of known concentration and semi-quantitation using a surrogate standard.
Formal Quantitation
In formal quantitation, a calibration curve is prepared using standards of the compound of interest prepared at various levels which bracket the concentration seen in the extract. Formal quantitation is the most accurate method and is favored for compounds which are known to be of unique concern (high toxicity) because this method is more accurate than the semi-quantitation method. However, a commercially available standard has to be obtained.
Semi-Quantitation
Semi-quantitation method employs a standard with a similar chemistry as compared to the unknown and is ideal when there is no commercially available standard. The method’s accuracy relies on the similarity of the instrument’s response for the surrogate standard as compared with that of the analyte of interest. Experimental verification can be very difficult and is hence assumed. Due to this reason, the use of the semi-quantitation method is not favored much.
Acceptable Levels for E&Ls
The acceptable level of the leachables is dependent on compounds and is assessed with the help of a toxicologist. The Product Quality Research Institute (PQRI) - an independent consortium from industry, the FDA, and academia have issued a guidance document for leachables and extractables for nasal and orally inhaled drug products. Within this context, they defined a safety concern threshold (SCT) of 0.15 µg per day for inhalation products.9 This can be subsequently transformed to an Analytical Evaluation Threshold (AET) to compare with the value established during an E&L analysis. The same quantity is used by the European Medicines Agency (EMA) as is target threshold. Other sources propose using a limit of 1.5 µg per day.11, 12
Quality Control in an E&L Study
Stringent quality control measures should be used to confirm the reliability and accuracy of an E&L study. Based on the quality system requirements for an individual E&L study, this includes some or all of the topics discussed below:
Analysis Blanks
All reaction vessels and solvents provide some level of background. The blank is utilized to corroborate the source of these components as well as to show that they do not originate from the sample. The analysis blank is a control sample which has been subjected to all the same steps utilized for the samples but which includes only the pure extraction solvent. The blank should be placed into the same type of extraction vessel and exposed to the same conditions just like the samples. Also, consideration should be given to enclosures which are likely to contribute to the background of the analysis.
Negative Control
Besides the analysis blank, other negative controls such as a known reference material or an instrument blank can be utilized to validate the cleanliness of the analytical system. The use of such controls can help establish that the systems utilized for the analysis do not contain extractable components as well as to show that a component did not originate from the sample. Instrument blank is the most commonly used negative control which shows that the analytical system does not contain any extraneous peaks.
Positive Controls or Spiking Study
The performance of the analytical method should be confirmed at the time of the analysis. A positive control refers to a reference material, which when examined shows that the method is working to expectations. A spiking study is utilized in one type of positive control. The spiking experiment estimates the accuracy of the method by adding a known quantity of target compound to the extract solution and indicating that an acceptable recovery is achieved. A commercially available standard should be available to conduct a spiking experiment. During E&L testing, spiking studies are particularly valuable because of the wide range of components that need to be quantified.
This places an additional burden on the analytical methods to be more generic and hence most of the compounds being studied have not been previously measured in the matrix of interest. The spiking study offers an effective way to verify the method accuracy for the target component.
Validation of Analytical Methods
Method validation is a process where a method’s performance characteristics are checked and confirmed to show that the method is indeed appropriate for the intended purpose. It is important to validate analytical methods as appropriate with regard to various parameters such as linearity, accuracy, limit of quantitation, limit of detection, range, specificity, system suitability and ruggedness. Most of these parameters apply to quantitative methods because these parameters can only be cross-checked when a specific compound is being analyzed. Also, validation of qualitative methods is more generic where a set of example compounds is used to show the methods suitability for a wide range of analytes.
Conclusion
An E&L study represents an integral part of validating the safety of a medical product. Moreover, design of an E&L study depends upon a good understanding of the materials used to develop the device as well as the expected use conditions. The best way to conduct E&L studies is to apply an analytical strategy which is informed by expectations of prospective extractables and which has adequate scope for discovering unanticipated components. The lab which performs the study is required to have adequate expertise in unknown identification to properly exploit information from multiple techniques, control experiments and databases to enable positive identification of unknowns. The lab should also have the required instrumentation to analyze a host of potential analytes. Finally, it should be an expert in analytical method development so that it can apply this knowledge to develop quantitative methods for the identified components.
Jordi Labs deals in the analysis of plastics and has the required knowledge and experience to make customers’ E&L study a success. The company partners with its customers to develop and perform E&L studies that are supported by more than three decades of analytical experience and sophisticated instrumentation.
Works Cited
1. ISO 10993-12 2012
2. BPA - https://www.fda.gov
3. Phthalates - Guidance for Industry Limiting the Use of Certain Phthalates as Excipients in CDER-Regulated Products
4. Corredor C, Tomasella FP, Young J. “Drug interactions with potential rubber closure extractables. Identification of thiol-disulfide exchange reaction products of captopril and thiurams,” J Chromatogr A., 2009, 1216, (1), 43–48
5. Guidance for Industry and FDA Staff - Total Product Life Cycle: Infusion Pump - Premarket Notification [510(k)] Submissions
6. Guidance for Industry and FDA Staff: Saline, Silicone Gel, and Alternative Breast Implants
7. Guidance for Industry and Food and Drug Administration Staff : The Content of Investigational Device Exemption (IDE) and Premarket Approval (PMA) Applications for Artificial Pancreas Device Systems
8. 21 USC § 351 - Adulterated drugs and devices, Section (a) 3
9. Safety thresholds and best practices for extractables and leachables in orally inhaled and nasal drug products
10. Mcneil Consumer Healthcare Announces A Voluntary Nationwide Recall Of All Lots Of Tylenol® Arthritis Pain 100 Count With Ez-Open Cap
11. Federal Register, Volume 60, No. 136
12. Ball, D., Norwood, D., Stults, C., Nagao, L., "Leachables and Extractables Handbook: Safety Evaluation, Qualification, and Best Practices Applied to Inhalation Drug Products," John Wiley & Sons, 2012.

This information has been sourced, reviewed and adapted from materials provided by Jordi Labs.
For more information on this source, please visit Jordi Labs.