Sponsored by ProtoJan 5 2021
When a sample fluoresces during a powder x-ray diffraction (XRD) experiment, the resulting errors in the quantitative results may render the experiment meaningless. Fluorescence is especially common in measurements performed with a copper x-ray tube on samples that contain iron.
Many tout the use of diffracted beam monochromators or energy-discriminating detectors as means to clean up the signal, but sample fluorescence indicates that microabsorption effects will be present – and microabsorption almost invariably causes errors in the results.
Fluorescence occurs when incident x-rays from the source excite the atoms of the sample, causing them to emit their own characteristic x-rays.

This effect is most pronounced when the source K-alpha radiation energy is only slightly greater than the characteristic x-rays of the sample. Unfortunately, the greatest effect coincides with the greatest difficulty in distinguishing the fluorescence x-rays from the source x-rays, as they are not very different in energy. Because of this large intensity of x-rays with similar energy, diffraction patterns tend to feature a greatly elevated background.
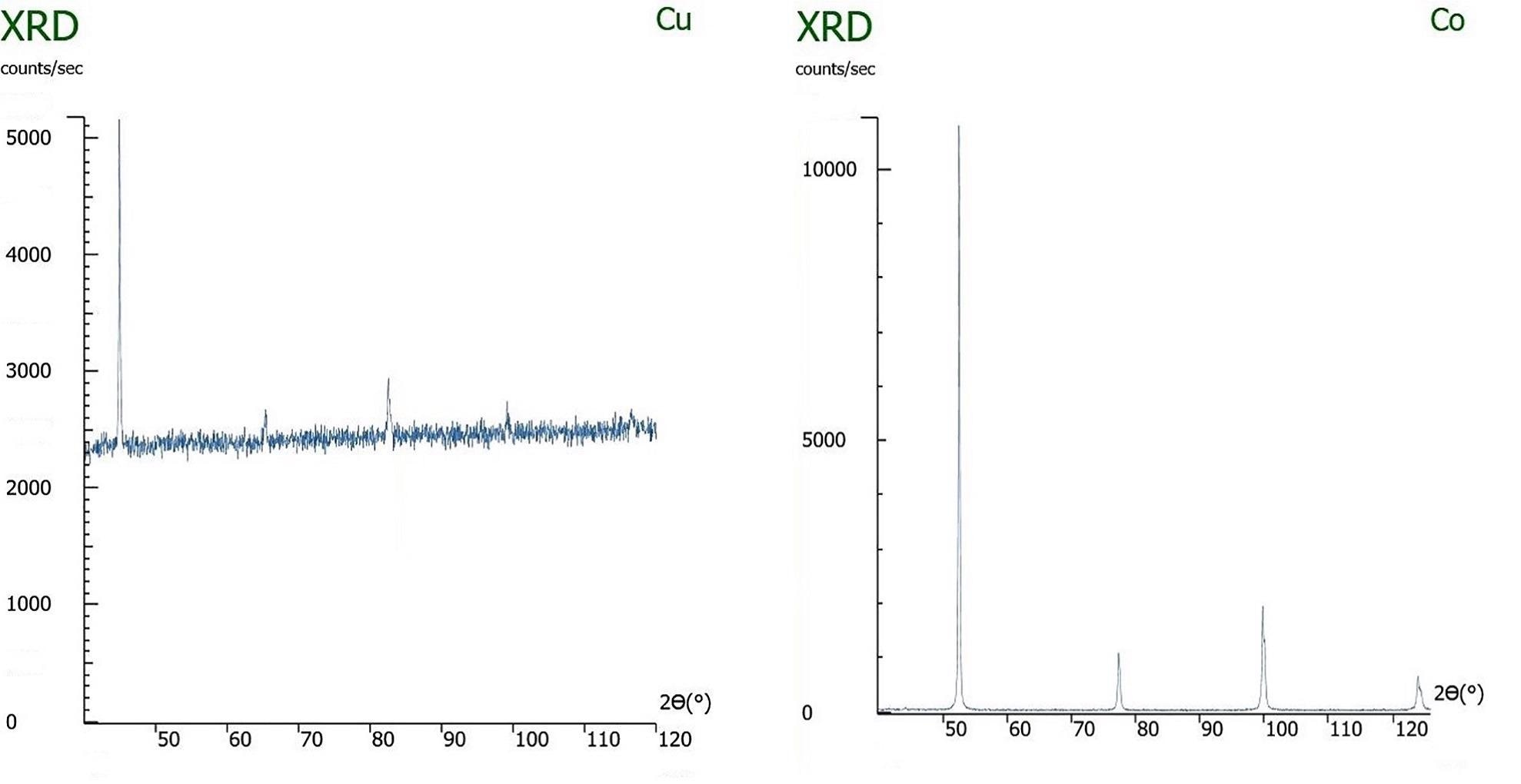
Left: An x-ray diffraction pattern of iron being measured with a copper x-ray tube, causing sample fluorescence. Right: The same sample being measured with the correct x-ray tube, cobalt, and not exhibiting fluorescence.
Diffracted beam monochromators or high energy resolution detectors can distinguish and exclude the sample fluorescence, thus greatly reducing the background, but they do not mitigate the effects of microabsorption. When the source radiation is most effective in stimulating fluorescence in the sample, the absorption of the source radiation by the sample will be most effective. If the sample has multiple components – which it will, when performing quantitative phase analysis – and they differ in their x-ray absorption constants, then microabsorption is very likely to drastically skew the apparent composition.
The significance of microabsorption in a sample depends on both the particle size (D) and linear attenuation coefficient (μ) of its component phases. The linear attenuation coefficient represents the fraction of incident x-ray beams that will be absorbed (or attenuated) per unit thickness. According to the Brindley criterion (not correction), for reliable quantitative analysis by XRD, the μD value should be less than 0.01. On the other hand, if the μD is higher than 0.01, a large fraction of x-rays will be absorbed by the material instead of being diffracted, causing that component to be underrepresented in the results. It is important to remember that in mixtures with multiple phases, the relative intensities of diffraction peaks may change depending on particle size if there is a significant difference between the linear attenuation coefficients of the elements or compounds.
Although the Brindley criterion can help determine whether a sample will exhibit microabsorption effects, there are challenges associated with this approach. Firstly, it is often difficult to quantify particle size, thus making it impossible to know the value of μD. With only a sieve available at many labs, there is no precise way to measure particle size. Secondly, even if particle size can be identified and altered via grinding, some materials require an extremely small particle size in order to achieve a μD below 0.01. For example, the particles may need to be ground to be smaller than one micron. In these situations, it is unlikely that the necessary equipment will be available to reduce particle size – and to verify the reduction – sufficiently, causing the μD value to be greater than 0.01.
On solid samples, the issue of microabsorption is even more difficult to mitigate, as the grain size will dictate the size of the crystallites. Thus, if the sample is not extremely fine grained and μ is not similar for all phases, the material will not meet the Brindley criterion, resulting in errors in practically any quantitative analysis.
To analyze the effects of fluorescence and microabsorption, iron and nickel were mixed in an equiatomic ratio. The sample was tested with both a copper and a cobalt source after being subjected to a range of grinding times. As shown in the graphs below, when tested with the copper x-ray source, which induces copious fluorescence in iron, the composition of the iron-nickel mixture was misrepresented: without any grinding, the indicated atomic fraction of nickel was approximately 70%, while the atomic fraction of iron was 30%. When the sample had been ground for an hour, the measured atomic composition was much closer to the actual composition, with an apparent nickel composition of about 55% and iron 45%. However, when the x-ray source was switched from copper to cobalt, which induces less fluorescence in iron, the XRD-measured composition of both elements was accurate even without any grinding.
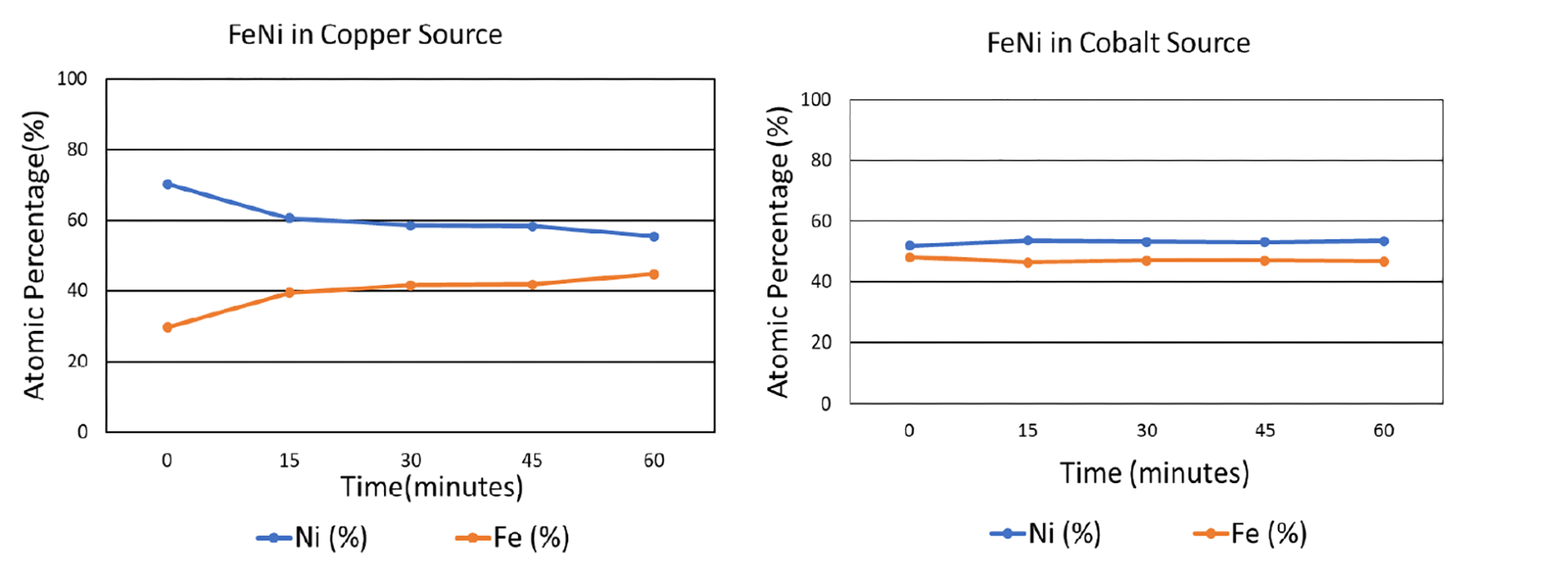
Based on these results, it is clear that choosing a compatible x-ray tube can be a highly effective solution in reducing the effects of microabsorption. This is especially important when (1) there is high background noise, (2) components being measured have very different linear attenuation coefficients, (3) a component being measured has a very high linear attenuation coefficient, (4) linear attenuation coefficients of components are not known, (5) missing peaks are observed, or (6) standardless analytical methods (e.g., Rietveld) are being used.
It should be emphasized that high background levels are not the problem. Rather, they indicate the likelihood of large μ, and thus μD, and microabsorption-induced inaccuracies. Reducing the background with monochromatization or energy discrimination will not improve inaccuracies caused by microabsorption. Therefore, the only guaranteed solution is to change the x-ray tube.

References:
[1] G. W. Brindley, Philos. Mag., 36, 347–369 (1945).

This information has been sourced, reviewed and adapted from materials provided by Proto.
For more information on this source, please visit Proto.