With the continued proliferation of Advanced Therapeutic Medicinal Products (ATMPs), therapy development, and marketed products available, the modern manufacturing process must adhere to the appropriate regulatory requirements.
In simple terms, ATMPs are pharmaceutical products or drugs; to modify existing ATMPs the cellular product is required as simple transplantation is not feasible. Where it is required that a more complex cell therapy should be manufactured, or when using cells in the treatment of a different organ, the processes must comply with regulatory agency approval standards. This ensures that manufacturing is conducted in facilities that present little to low (or acceptably low) patient risk.
The FDA and EMA hold the responsibility for ensuring the safety, efficacy, and compliance with Good Manufacturing Practices. Included in the GMP requirements, among other things, is ensuring there is adequate environmental monitoring in critical areas and accounting for special requirements that ATMP production demands.
Employing adaptable standard environmental monitoring technologies makes it easy to modify systems to meet ATMP production requirements. Where possible, the implementation of new, sophisticated technologies bolsters the contamination control strategy for the overall risk management of production environments and workflows.
Overview of Process
Preparation of common ATMP products differs from conventional biopharma product manufacturing as it is produced from the initial cells collected from the recipient patient. Before returning the cells to the donor patient, they are modified per therapy type.
ATMP batch volumes are small scale as the process and product are tailored to the needs of an individual patient, they also do not make use of sterilization steps that could cause damage the final cellular product. This means that the entire process flow must be aseptic to avoid corruption of the cell batch, and crucially, to prevent cross contamination between batches.
Gene therapy differs slightly in that cells obtained from a single patient can be modified to meet the therapeutic needs of a greater population of patients, and although larger than individual cell therapy batches, batch sizes remain relatively still small in scale in relation to classic pharmaceutical drug manufacturing. Through each stage of the process, these small batches are typically manipulated by hand making the contamination risk more pronounced. By using isolator glove boxes, external contamination risk factors are reduced but it can make manipulations more complex; as scale increases, robotics are being considered to move the whole process to a ‘single box’ manufacturing format.
Environmental Monitoring
The central importance of monitoring environmental conditions in ATMP facilities is to ensure the highest degree of control is maintained over the following two processes:
- The aseptic manufacturing environment
- The potential for cross contamination between batches/manipulations
These two primary processes, are considered open processing and closed processing environments. When performing open system manufacturing, the processes must be compliant with Grade A (ISO5) while maintaining unidirectional airflow withing Grade B (ISO7) room environment standards. These ambient environments are at an increased risk of contamination as operators (the primary source of contamination) are withing the vicinity of the processing being performed; operators must perform routine functions within the environment. Conversely, closed processing isolates all conditions from the processing environment, and in order to perform manipulations, operators must use glove ports fixed within the isolator. CIP/SIP processing can also be automated using isolators.
Environmental Monitoring to Demonstrate Aseptic Control
Regulatory guidance offers guidelines when it comes it best practices for monitoring processes in aseptic manufacturing. The EU GMP Annex 1 (updated and released recently in August 2022) provides details on how to assert control over the aseptic environment, specifically the Grade A critical areas. Rigorous monitoring of the environment is crucial to ensure that there a complete and consistent awareness of ongoing conditions, including the detection of periodic events which could be disastrous if undetected.
Through persistent monitoring, a continuous flow of information is generated which results in in a vast quantity of data which can be used to observe potential trends. Therefore, the manufacturing facility implements an extensive environmental monitoring program. This should include monitoring for viable and nonviable airborne particulates, surface viable contamination, and personnel, in the aseptic areas.
These procedures should establish the required frequencies and locations for the monitoring sample points, implement warning and alarm limits for each area, and assert corrective actions if any of the areas deviate from the expected results. When limits are exceeded, the appropriate action includes an investigation into the source of the problem, while evaluating the potential impact on the product and subsequent measures required to prevent a recurrence.
A Contamination Control Strategy (CCS), which includes an environmental monitoring program, should be implemented facility wide. As part of a risk assessment, the CCS should determine key control points and evaluate the effectiveness of the controls and monitoring measures implemented for the risk management associated with contamination.
Monitoring should be conducted using the appropriate methods that comply with any Risk Assessment standards. Continuous monitoring of Grade A areas (for particles ≥0.5 and ≥5 µm) is required and should account for a suitable sample flow rate (at least 28.3 LPM / 1 CFM). This ensures all interventions, transient events, and system deterioration are captured.
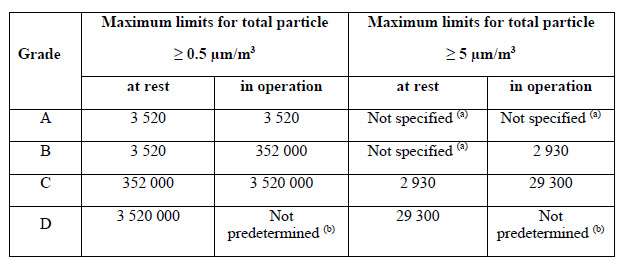
Figure 1. Annex 1 particle limits table. Image Credit: Particle Measuring Systems
The system should routinely make a comparison of each individual sample result with alert levels and action limits. The frequency of this action should be habitual so that any potential excursion can be determined early on for a timely response – when alert levels are exceeded, Alarms should be triggered.
Procedures should determine the appropriate actions in response to alarms including the recognition of additional microbial monitoring. Dedicated point of use sensors helps satisfy the requirement for continuous monitoring within Grade A areas; these sensors are networked via a central monitoring software application that can alert operators within the cleanroom or messages to relevant groups with the appropriate alarm outputs. A permanent record of these alert and alarm excursions should be recorded in the audit trail of the system.
Risk Assessment – Part 1
One feature of the system is sample point location; this should be established in accordance with a documented Environmental Monitoring Risk Assessment (EMRA) and should include the following information:
- Sampling locations
- Frequency of monitoring
- Monitoring method used
- Incubation conditions (e.g., time, temperature(s), aerobic and/or anaerobic conditions).
The risk assessment is determined by inputs provided by the different groups across the facility.
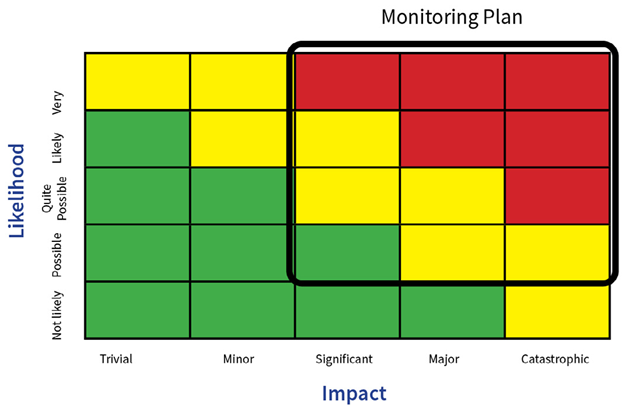
Figure 2. Monitoring plan. Image Credit: Particle Measuring Systems
Typical Automated Continuous Monitoring System
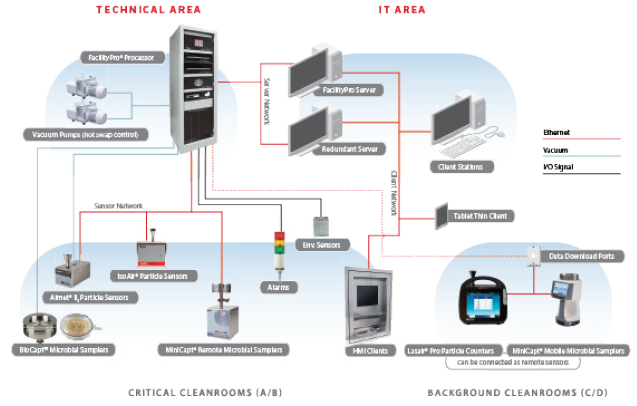
Figure 3. Schematic of theoretical FacilityPro configuration. Image Credit: Particle Measuring Systems
When constructing an integrated solution, the instrumentation used will typically include:
Particle Counting: Dedicated sensors at each location are required for continuous data acquisition. These sensors should continuously sample throughout the assembly and production phases of manufacturing. Sample points should be situated within the hood or isolator line and attached to the sensor via a short length of sample tubing (no more than 2 m, preferably shorter). The risk assessment outcomes determine the location and orientation of the probe.
Microbial Sampling: Where a need for particle counting has been established through risk assessment, there is an corresponding need to conduct microbial sampling.
Active Air Sampling: To ensure that samples are not exhausted locally within the critical space, only the sample head is to be placed within the environment. The sampling is quantitative and can continuously run for up to 4 hours. The software interface is used to control start and stop commands.
Static/Passive Air Sampling: For a period up to 4 hours, a plate is positioned in the local environment providing an additional data point valuable to gaining an overall understanding of the microbial risk. Certain microbiologies can be damaged by the active nature of impaction sampling, and although it is not mandatory to include this method as part of routine monitoring, it should form an aspect of qualification studies and the EMPQ.
Rapid Automated Microbial Monitoring (RMM): The use of autofluorescence microbial instrumentation, with the capacity to differentiate inert and biological particles in real-time, introduces new element to determine how control within the environment is demonstrated. Alongside total particle and active and passive air sampling, it provides extra value in making rapid decisions relative to loss of control within an area. Demonstration of asepsis and sterility is not guaranteed by a single piece of evidence; this is outlined the introduction to the environmental monitoring chapter in EU GMP Annex 1.
Environmental & Process Monitoring 9.1
“The site’s environmental and process monitoring programme forms part of the overall CCS and is used to monitor the controls designed to minimize the risk of microbial and particle contamination. It should be noted that the reliability of each of the elements of the monitoring system (viable, non-viable and APS) when taken in isolation is limited and should not be considered individually to be an indicator of asepsis. When considered together, the results help confirm the reliability of the design, validation and operation of the system that they are monitoring.”
The accumulation of data and information offers a comprehensive understanding of the effectiveness of the established environmental controls. For more than two decades, the convergence of particle and microbial data has been traditional platform for demonstration of environmental control, where ideally no ≥ 5 µm would exist. This was due to large particles being deemed as having the potential to transport agents for microbiology or being clusters of microbiological entities with the capacity to sustain viability in the dry air associated with cleanroom environments. Once conventional microbiology and RMM provided the evidence of the presence of those particles in particle counting, the intersection of the three areas of data is where the relationship of potentially harmful ≥ 5 µm particles may observed, microbiologically and in real-time.
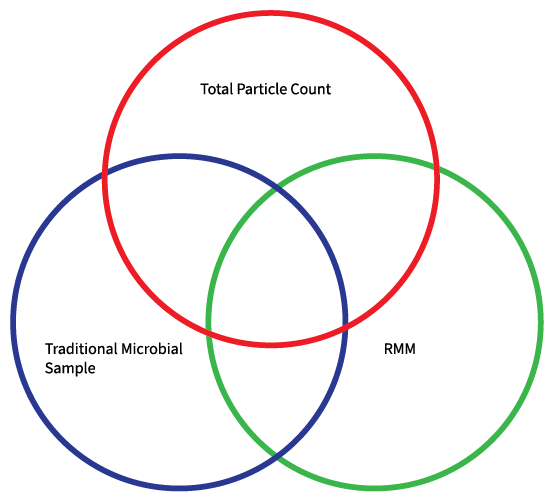
Image Credit: Particle Measuring Systems
Environmental Monitoring to Demonstrate Cross Contamination Control
The strength in a program’s design which can demonstrate the capacity to reveal cross-batch contamination, or intra-batch contamination, is dependent on the kind of processing being carried out. Several facilities advocate for the use of dedicated rooms per batch during its processing stages and filling.
Each room must include a central bio-safety hood within a Grade B background. The responsibility for maintaining the preparation, gowning, cleaning, and environmental monitoring required in each space lies with the operators. Sample point location(s) should reflect the various activities conducted within the hood/room, and the flexibility in design of the monitoring system is a necessity.
Upon completion of a process, preparing the space for the next batch of product should be performed in the appropriate manner. The environmental monitoring conducted throughout the entire run of each batch demonstrates separation and the effectiveness of the cleaning and sanitization protocols.
Continuous monitoring systems use tags in the data to distinguish between each batch record without having to insert a pause in monitoring. This facilitates easy transitions between phases of the process; distinct recipes related to each phase can allocate the proper alert and action limits specified for those activities.
Where a production line process is fixed in place for cleanroom operations (i.e., where one product manipulation is conducted at a single stage and then advanced along the process for subsequent steps), the environmental sample probe location can be adjusted to better align with the process. Each step can be continuously monitored, and during the ‘at rest’ phases between batches, bar codes can be used as markers for unique data identifiers.
At the end of the processing run, a final report can be generated to show the progress and rest phases. Where processing takes place in the various modules of an isolator, operators can sanitize between batches, if required. Conversely, both sanitization and surface monitoring can help meet compliance when performed routinely.
For QbD operations, validation of the process can be achieved as tags in the data can display the particular functions for intra-batches and the system is not as dependent on finished product testing as may have been established previously. Optionally, there is a third hybrid operation that groups functions into those conducted in lower grade zones and those that must be performed under unidirectional flow. The tracking of this application data in these scenarios becomes an important parameter for reporting. Batch identification tags within the data (bar codes, RFID, etc.) make the reporting of the finished product easier. Intra-batch isolation becomes concentrated on critical risk activities, and continuous monitoring is achieved throughout.
Environmental Data Reporting
Data and Status Information Displays: A central reporting tool is utilized due to the collected data being multivariate. By combining the data from the different environmental components, alongside the tagging of product during production, enables a clear visualization of the facility layout: this includes dashboards for current data and status information for each batch in relation to the room and stage in real-time. It is also possible to view data, status, and sampling information for each dedicated area on a single screen.
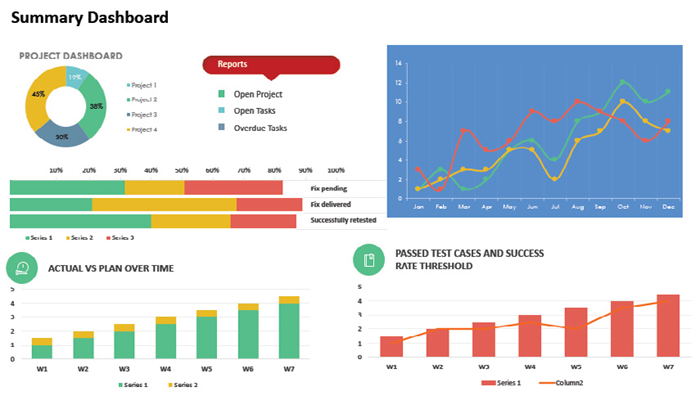
Image Credit: Particle Measuring Systems
Report Generator: A capable data report generator is required to provide legible reports for all stored data: audit trails (events), data/statistical summaries, and trend charts. The system should have historical data retrieval capabilities (as determined in the site User Requirement Specification) for the associated system. To support the release of product, filters for data, time, location, and batch, should make the data easily to access and, when required, easy to export, or print.
Image Credit: Particle Measuring Systems
Alarms: Programming alarms in line with the product and process steps can be monitored and reported via the dashboard interface with the capacity to sort alarms by different criteria. The display shows an alarm acknowledgment function; date, time, area, and description, as well as any additional information needed.
Image Credit: Particle Measuring Systems
Alarms programmed to reflect any trends must involve a review of the process to establish any increasing excursions from action limits or alert levels. Where any frequent or consecutive excursions are observed a common cause may be the root problem. For microbial limits, it is crucial to take into account not only the quantity of CFU detected but also any shifts in qualitative information, including type and the predominance of certain organisms.
Risk Assessment – Part 2
The information obtained in processing environmental data should be resubmitted into the production activities. Review of operations should be conducted when specific functions give cause for an environmental concern: increase of baseline values increasing towards alert or actionable thresholds. The review shall consist of a gap analysis that compares the initial risk assessment against a review of the control point efficacy. This must take into account any recommendations or changes for improvement or enhanced/additional monitoring to mitigate any potential functions that may cause defects in the future.
Summary
A monitoring system which continuously scans for environmental parameters (including total particle, traditional microbiology, rapid microbiology, air velocity, temperature, humidity, air exchange rates, etc.) will determine how a cleanroom should be maintained to meet operation standards.
By associating the corresponding data with the batch, it can be demonstrated that intra-batch separation is established and maintained. The monitoring and recording of sanitization help complete the batch record. All the relevant data can be visualized on a central dashboard facilitating precise, rapid analysis.

This information has been sourced, reviewed, and adapted from materials provided by Particle Measuring Systems.
For more information on this source, please visit Particle Measuring Systems.