Biopolymeric filamentous networks are key components in the living world. Some key examples include the connective tissue (collagen), the cell cytoskeleton (actin), and the blood coagulation system (fibrin).
These processes are a result of the polymerization or assembly of macromolecular monomers, and can be examined with time-resolved scattering methods triggered by combining pure components. The application of a DAWN DSP MALS instrument was previously shown to demonstrate the early reaction stages of the fibrinogen (FG) assembly following enzymatic activation, recovering the time evolution of the <M>w and <Rg2>z of the ensembles.
It is not possible to recover the evolution of fiber cross-section - a key structural parameter in fibrous structures formation - in the early reaction stages from MALS data. This fiber cross-section could be determined by the so-called Casassa plot method, but most reactant species are too short (< 200 nm) to enable its application. Alternatively, the cross-sectional mean square radius <Rc2>z can be recovered from SAXS data.
An experimental set-up containing a SAXS flow-through capillary cell, a Peltier-equipped HELEOS-II 18-angles MALS detector, and a stopped-flow mixer was assembled at the SWING beamline of the synchrotron SOLEIL. The MALS detector plays a key role in this set-up, which helped in following the combined evolution of <Rc2>z versus <M>w and <Rg2>z versus <M>w during the early phases of fibrin polymerization.
Modeling potential reaction pathways revealed that fibrin assembly’s traditional mechanism, involving the instant formation of double-stranded fibrils with a half-staggered assembly of monomer units, was not sufficient to elucidate the data.
A revised model was suggested that is fully compatible with the data, and could also clarify other aspects of the fibrin network.
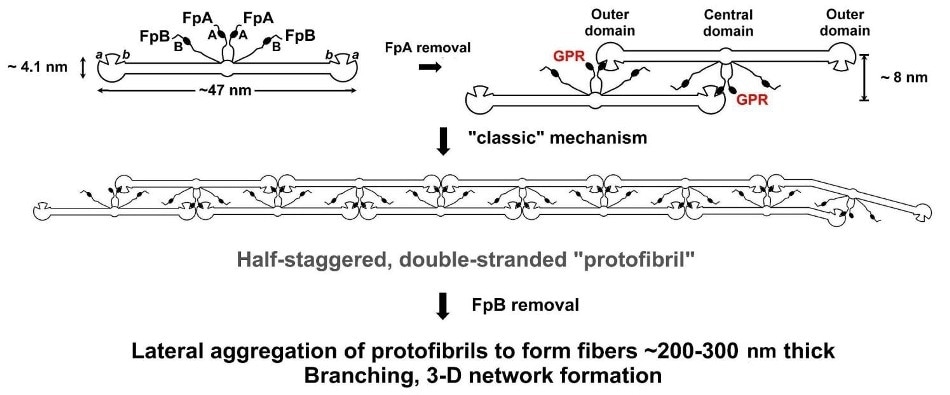
Figure 1. The “classical” model for fibrinogen network formation. Following enzymatic removal of two short peptides (FpA) from the central domain of FG molecules, Gly-Pro-Arg (GPR) "knobs" are uncovered, which are complementary to "holes" always present in the outer domains of every FG molecule. This initiates self-assembly, producing long, half-staggered, fully double-stranded (proto)fibrils. Lateral fibril aggregation and branching aided by delayed enzymatic removal of another pair of peptides (FpB) generate the final fibrin network.
Materials and Methods
Reagents
Only reagent-grade chemicals were used for the experiment, and MilliQ water was employed for all of the solutions. Lyophilized, plasminogen-depleted human FG was obtained from ERL.
It was reconstituted at 20 mg/ml and purified across a preparative size-exclusion column prior to the experiments to create approximately 20 ml at about 2 mg/ml stock solutions. Sigma-Aldrich’s A-5042 (Ancrod activating enzyme from snake venom) was procured.
Vials with 50 NIH units/ml aliquots were prepared and kept at -80°C. GPRP-NH2 was H-1998 from Bachem.
For all experiments, NaCl 104 mM, Tris 50 mM, aprotinin 10 KIU/mL, pH 7.4 (TBS), with or without CaCl2 1.25 mM, were used as buffer.
Instrumentation
Remote-controlled apparatus is required to make SAXS beamline measurements. The entire set-up comprised of a four-syringe, stopped-flow mixer altered with the integration of a mixer, external solenoid valves, and a splitter.
This set-up prevents cross-contamination during prolonged waiting times between injections and enables rapid flushing of the external mixer once the injection has been completed. An in-line, 70 µl PEEK/titanium 0.1 µm filter housing is located immediately after the mixer to eliminate particulates.
![Schematic diagram of the stopped-flow MALS/SAXS set-up. Two positions of the lower valve are shown; other valve combinations are used during an experiment (see [5]). Green, buffer; magenta, reacting mixture.](https://d12oja0ew7x0i8.cloudfront.net/images/Article_Images/ImageForArticle_13105_45065256888472226474.jpg)
Figure 2. Schematic diagram of the stopped-flow MALS/SAXS set-up. Two positions of the lower valve are shown; other valve combinations are used during an experiment (see [5]). Green, buffer; magenta, reacting mixture.

Figure 3. The combined MALS-SAXS apparatus in the SWING beamline hutch at SOLEIL.
The reaction mixture should not be disturbed within the MALS cell for time-resolved measurements, but it should be regularly refreshed while in the SAXS capillary. This is done to ensure that the strong X-ray beam does not cause any irreversible damage to the protein.
A four-way rotary valve, located between the SAXS and MALS cells, separates the MALS cell from the rest of the apparatues soon after injection and directs the flow to the SAXS capillary. Solution was pushed via the SAXS capillary in increments of 5 µl every two X-ray shots, with each X-ray shot lasting about 0.5 seconds with an interval of 5 seconds. Approximately 1.6 ml was needed to fill the cells as well as the accumulation loop.
MALS Data Acquisition and Analysis
The HELEOS-II cell was stabilized at 20±0.1°C to ensure that the static MALS sample is not considerably heated by the laser. Using the ASTRA 6.0.3 software, initial analysis and data acquisition were carried out.
A dn/dc value of 0.192 cm3/g was employed for FG and for TBS, n =1. 3332 at λ= 651 nm, making 16 scattering angles available, between 13.5° and 157.5°. The QELS fiber optic was located in position #12 at approximately 100°, and using the Rg of FG, which was directly determined by SAXS on the same sample, normalization was carried out.
In the same file, more than a single polymerization run was obtained, directly injecting the buffer-mixture-buffer sequences while the ASTRA software was running. Figure 4 shows an example of a partially processed run.
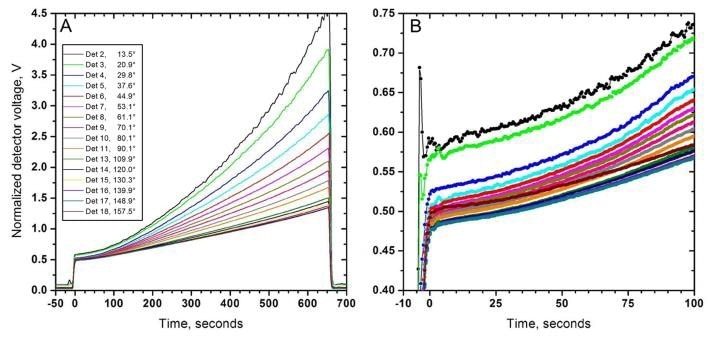
Figure 4. Angular MALS data (normalized voltages) collected by a HELEOS-II from a 0.46 mg/ml fibrinogen (FG) sample undergoing polymerization induced by 0.05 NIH units/mg FG of the snake venom enzyme "Ancrod". The reaction was stopped before the gel point. Panel A, complete dataset. Panel B, the first ~100 s on an enlarged scale. Times are rescaled to the end of the injection phase. Note the excellent quality of data even at the lowest angles.
The Zimm formalism was used to process the data. As the reaction creates a heavily polydisperse ensembles of rod-like species, the Zimm plots deviate quickly from angular linearity.
Each dataset was equipped with varying polynomials from the 1st to the 5th degree, excluding and including the two lowermost angles (Figure 5). The resulting <M>w and <Rg2>z datasets were subsequently sent to an Excel® spreadsheet to physically collate sections for the best possible fitting of angular dependence and eventually establish <Rg2>z values (<M>w values being less affected by the specific fitting process).
More simulations were carried out to establish that this process accuracy provides the <Rg2>z values of polydisperse mixtures of rod-like particles.
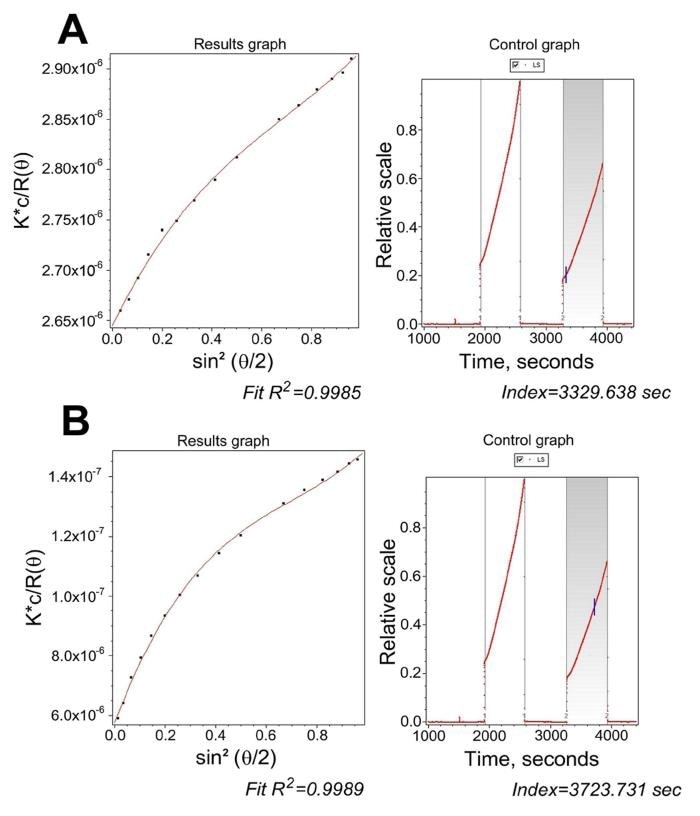
Figure 5. ASTRA processing of the data from Fig. 4. Panel A, a slice collected at 51 s after mixing, highlighting the need for no less than a 3 rd degree polynomial and for including the 20.9° scattering angle. Panel B, a slice collected at 445 s after mixing, where a 5th degree polynomial was required using all angles.
SAXS Data Acquisition and Analysis
The distance of the SAXS detector was 4 m for the nonpolymerizing FG runs. Following in-house data treatment, <Rg2>z values were acquired from cross-section Guinier plots with the US-SOMO SAS module.
Modeling
A complete description of the complicated fibrin polymerization modeling, which uses an altered Flory bifunctional polycondensation scheme and indicates individual FG units as a cylinder, is described in Rocco M, et al. 2014. J. Am. Chem. Soc. 136, 5376-5384.
The modeling allows the release ratio Q between the two FpA from a FG molecule to be accounted for. Distributions produced by high Q values are skewed toward long polymers.
Results and Discussion
Figure 6 shows a sequence of datasets plotting the weight-average molar mass <M>w, mean square radius <Rg2>z , and mean-square thickness <Rc2>z versus time for FG polymerizations caused by Ancrod under varying solvent conditions — BS, TBS + CaCl2 1.25 mM, TBS + GPRP-NH2 in a 10-fold molar excess to FG.
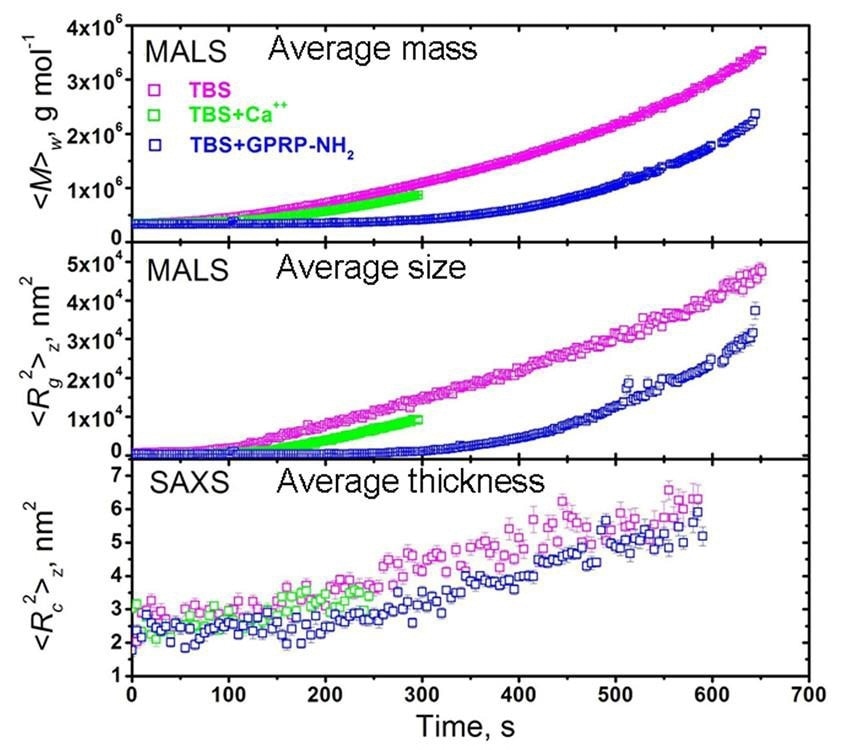
Figure 6. Coupled MALS/SAXS measurements of activated fibrinogen polymerization under various conditions: TBS, is a Tris saline buffer; TBS with Ca++; and TBS with GPRP-NH2, a peptide that competes for the binding sites, used in rate-limiting amounts. Reactions were stopped before the gel point, which occurs earlier with Ca++ .
Large variations were seen between the three conditions in the time evolution of <M>w and <Rc2>z , while only a mild variation was observed in the <Rc2>z data when the GPRP-NH2 inhibitor is present in comparison to no GPRP-NH2.
Conversely, after removing the time factor from the same data by plotting <Rc2>z versus <M>w (Figure 7A), the three datasets overlay on each other, demonstrating that the fundamental polymerization mechanism is the same, regardless of the huge difference in the time evolution.
A slight difference was seen in the <Rc2>z versus <M>w plot when the GPRP-NH2 inhibitor exists in rate-limiting quantities.
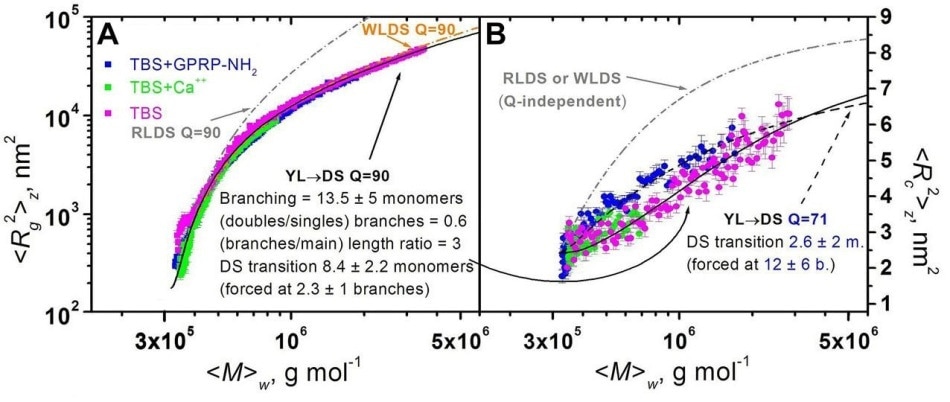
Figure 7. Plots of mean square radius vs. weight-average molar mass (A) and of mean square thickness vs. weight-average molar mass (B) for FG polymerizations under different conditions. Superimposed are model curves generated for some polymerization models (see text).
Modeling was subsequently performed to infer the results. Initially, the traditional model was used.
Fully half-staggered, rod-like double-stranded (RLDS) rigid polymers could fit only the initial part of the <Rc2>z versus <M>w plots (indicated as a grey dashed curve in Figure 6A). Fully half-staggered, worm-like double stranded (WLDS) semi-flexible polymers were able to fit the whole the <Rc2>z versus <M>w datasets (indicated as an orange dashed curve in Figure 7A).
However, these models were not able to fit the <Rc2>z versus <M>w curves (indicated as dashed grey curves in Figure 7B), demonstrating that the width of the polymers gradually increases than that expected by the traditional model.
Following this, a new fibrin polymerization model was developed, involving one binding event and a deferred transition to the fully double-stranded (DS) fibrils, as shown in Figure 8A.

Figure 8. The "Y-ladder to double-stranded” (YL → DS) model for fibrin polymerization. Panel A, the single-binding to full DS transition. Panel B, top: branched structures generation by off-axis binding of a growing chain to a still available A knob; bottom: a typical resulting branched fibril, with the growing ends still in the YL configuration.
This elongation mode creates polymers, whose Rg is close to that of their equivalent fully DS entities and also whose cross-section, as viewed by SAXS, is the same as the parent FG monomers.
In addition, the new model enables branches to grow at an early stage (Figure 8B). By adjusting the DS transition and the occurrence of branching, it was possible to create model curves fitting the <Rc2>z versus <M>w as well as the <Rc2>z versus <M>w plots (Figure 7, black lines).
Conclusion
During the initial stages of a polymerization reaction, fibrin networks show the formation of rod-like, elongated monomers and then proceed to the long fibril assembly that ultimately branch and thicken. This complex behavior can be described by MALS measurements.
The evolution of shape- and size-related parameters is established by time-sequenced MALS data. The optimal angular range along with the consistent data, rendered by the temperature-controlled 18-angle DAWN instrument, form a strong foundation for comprehensive modeling studies needed to elucidate such systems.
The concurrent SAXS data acquired, together with the MALS data, acquired through the DAWN’s flow cell configuration, was important to determine the fibrils’ lateral growth profile as they branch and thicken.
The standard model of fibrin network formation does not promote early branching during the polymerization process. This paradigm was confidently revised based on the strong results of MALS-SAXS analysis.
In the new model, a few branches in the growing fibrin lumps may possibly interconnect, while other branches collapse on the newly-formed network filaments, resulting in thicker yet low-density fibers.
The ASTRA software offered sophisticated capabilities for the acquisition and processing of MALS data, but the complex, polydisperse nature of the mixtures of rod-like particles formed in the fibrin polymerization process stipulated the need for further data treatment. Consequently, a manual method was developed to detect the polynomial order and optimal angular range for studying nonlinear Zimm plots. Work is being done to automate this task to some extent.
The rapid-mixing MALS/SAXS set-up described in this article has generated high-quality data. Researchers analyzing similar reactions, creating filamentous networks in the synthetic or biological polymers fields can exploit this new opportunity if they have already defined their systems with their own specific MALS instrument at their laboratories.

This information has been sourced, reviewed and adapted from materials provided by Wyatt Technology.
For more information on this source, please visit Wyatt Technology.