Introduction Recently, the preferential oxidation (PROX) of CO in H2-rich gas has attracted new interest due to its application in fuel cell technology. For instance, polymer electrolyte fuel cells (PEFC) are attractive for small scale and automotive applications, although PEFC requires high purity H2 as fuel with CO concentrations below 10 ppm since the anode Pt catalyst is easily deactivated by CO contamination [1, 2]. PROX reaction is one of the most straightforward method to reduce the CO contamination in H2-rich gas. For now, noble metals (Pt, Rh, etc.) supported catalysts have been tested and proposed for thermal PROX reaction [3-5]. On the other hand, highly dispersed transition metal oxides such as V [6], Cr [7, 8] and Mo [9-11] incorporated within the framework of zeolites or supported on various oxides show unique photocatalytic activity for various reactions such as decomposition of NOx, oxidation of CO and partial oxidation of hydrocarbons, under not only UV and/or visible light irradiation. However, to the best of our knowledge, no attempt has been made to apply photocatalysis for the PROX reactions. In the present work, highly dispersed Mo6+-oxide species supported on silica (Mo/SiO2) and Al2O3 (Mo/Al2O3) were prepared by an impregnation method and characterized by various spectroscopic techniques. Furthermore, the photocatalytic selective oxidation of CO into CO2 with O2 in the presence of H2 was investigated and compared with that of TiO2 (P-25) photocatalyst. Experimental Mo/SiO2 and Mo/Al2O3 (0.60 wt% molybdenum as metal; 62.5 μmol of Mo/g-cat) were prepared by an impregnation method of SiO2 (aerosil 300; Degussa) or Al2O3 (Aluminium Oxide C; Degussa), respectively, using an aqueous solution of (NH4)6Mo7O24∙4H2O. After impregnation, the catalyst was dried at 373 K for 12 h and calcined at 773 K for 8 h in air. TiO2 (P-25; Degussa) was calcined at 723 K for 8 h in air. Prior to the photocatalytic reactions and spectroscopic measurements, the catalysts were calcined in O2 (> 2.66 kPa) at 773 K (at 723 K for TiO2) for 1 h, and then degassed at 473 K for 1 h. Diffuse reflectance UV-vis spectra were recorded at 298 K with a Shimadzu UV-2200A double-beam digital spectrophotometer. The XAFS spectra were obtained at the BL-19B2 facility of the SPring-8 at the Japan Synchrotron Radiation Research Institute (JASRI). The synchrotron radiation from 8.0 GeV electron storage ring was monochromatized by a Si(1 1 1) monochromator. The Mo K-edge spectra were measured in the transmission mode at room temperature. Curve fitting analysis of the EXAFS spectra was conducted on k3χ (k) in k-space (k range = 3-12 Å-1) with a REX2000J program (Rigaku). The FT-IR spectra were recorded at room temperature with a FT-IR spectrometer (JASCO FT-IR 660 Plus) with self-supporting pellets of the samples in the transmission mode. The photoluminescence spectra were measured at 298 K with a SPEX Fluorolog-3 spectrofluorometer. Photocatalytic reactions were carried out in a closed system using quartz reactor (reaction volume: 101 cm3) under UV irradiation at 293 K using a 100 W high-pressure mercury lamp (Toshiba SHL-100UVQ-2) through water filter. In the present investigation, gas concentrations are defined as ppm, that is, the concentration of gas with its partial pressure of 1 atm is equivalent to the concentration of 106 ppm. The initial concentration of gasses was CO 920 ppm, O2 1815 ppm and H2 5950 ppm. The amount of the catalyst used for photocatalytic reactions was fixed at 50 mg. The reaction products were analyzed by on-line gas chromatography. Results and Discussion The photocatalytic selective oxidation of CO into CO2 with O2 in the presence of H2 was investigated on Mo/SiO2 and Mo/Al2O3. As shown in Figure 1, UV irradiation of Mo/SiO2 led to the efficient oxidation of CO into CO2, accompanying the stoichiometric formation and consumption of CO2 and O2, respectively. Concentration of CO gas reached below the detection limit of GC analysis (less than 8 ppm) after UV light irradiation for 180 min, while the amount of H2 remained constant. The turnover number for the reaction defined as the ratio of the amount of CO2 to the amount of Mo6+-oxide species included in the catalyst exceeded unity, indicating that the reaction proceeded photocatalytically. The amount of CO2 produced as well as the amount of O2 and H2 consumed reached 920 ppm, 460 ppm and 7.26 ppm, respectively. From these values, the CO conversion and selectivity were calculated to be ~100% and 99%, respectively, by using the following equation: CO selectivity (%) = {(CO2 (t=180min)) / [(H2 initial - H2 (t=180min)) + CO2 (t=180min)]} x 100 (H2 initial: the initial amount of H2, CO2(t=180min), H2 (t=180min): the amount of CO2 produced and the amount of H2 in the gas phase after the UV irradiation of 180 min). 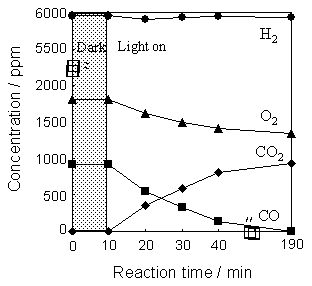
Figure 1. Reaction time profiles of the selective photocatalytic oxidation of CO with O2 in the presence of H2 on Mo/SiO2 under UV light irradiation at 293 K. Moreover, as shown in Figure 2, the time profiles of the amounts of CO, O2 and CO2 (depicted as solid line) show good coincidence with that of the reaction performed in the presence of H2 (Figure 1). These results indicate that the CO oxidation reaction is hardly affected by the presence of H2. In contrast to the Mo/SiO2, it was found that Mo/Al2O3 does not exhibit any photocatalytic activity for this reaction. Moreover, photocatalytic oxidation of CO with O2 in the presence of H2 was also performed on the TiO2 (P-25). CO oxidation reaction proceeds on TiO2, however, CO conversion (81%) as well as CO selectivity (89%) of TiO2 after UV irradiation of 360 min were lower than those of Mo/SiO2. Then, the photocatalytic oxidation of H2 with O2 were performed on Mo/SiO2 and TiO2. As shown in Figure 2, the amounts of H2 and O2 clearly decreased on TiO2 after UV light irradiation for 180 min (depicted as open symbol), however, the amounts of H2 and O2 hardly changed on Mo/SiO2 (depicted as dotted line). These results indicate that H2 is hardly oxidized by O2 on Mo/SiO2 under UV irradiation. 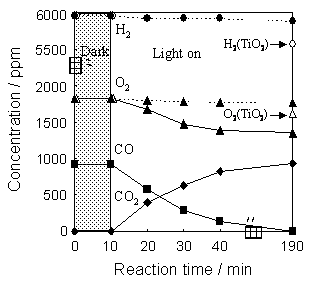
Figure 2. Reaction time profiles of the photocatalytic oxidation of CO with O2 on Mo/SiO2 (solid line) as well as the photocatalytic oxidation of H2 with O2 on Mo/SiO2 (dotted line) and TiO2 (open symbol) at 293 K. In order to elucidate the active sites for the photocatalytic reactions and their local structures, Mo/SiO2 and Mo/Al2O3 were characterized by various spectroscopic methods. The local structure of Mo6+-oxide species on SiO2 and Al2O3 were investigated by Mo K-edge XAFS measurements. XANES spectra of Mo/SiO2 and Mo/Al2O3 exhibit well-defined preedge peaks due to the 1s-4d transition of Mo atoms, indicating that Mo6+-oxide species exist in a tetrahedral symmetry for both samples (Figure 3 (A), (B)). The Fourier transformed EXAFS (FT-EXAFS) spectra of Mo/SiO2 and Mo/Al2O3 showed peaks due to the existence of neighboring oxygen atoms (Mo-O) at around 0.8-2.0 Å, while the additional peak due to the Mo-O-Mo bond was not observed between 3.0-4.0 Å. These results suggest that Mo6+-oxide species are highly dispersed on SiO2 and Al2O3 [12, 13]. Moreover, the results of curve fitting analysis of Mo-O bonds suggest that Mo6+-oxide species on SiO2 and Al2O3 have two short Mo=O double bonds and two long Mo-O single bonds, while Mo6+-oxide species on Al2O3 has longer M=O double bonds and shorter Mo-O single bond than that on SiO2, as shown in Table 1. The long Mo-O single bond of Mo6+-oxide species on SiO2 can be ascribed to the small ionic character of framework metal-oxygen bond (Si-O) of SiO2 as compared to that (Al-O) of Al2O3 [14]. The long Mo-O single bond also indicates the weak interaction between Mo6+-oxide species and SiO2 [14]. 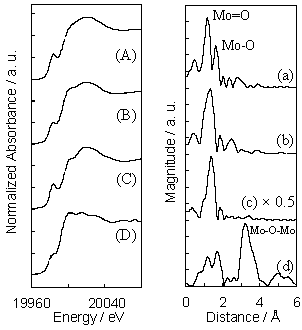
Figure 3. XANES (A-D) and Fourier transforms of EXAFS (a-d) spectra of: (A, a) Mo/SiO2, (B, b) Mo/Al2O3, (C, c) Na2MoO4 and (D, d) MoO3 Table 1. Results of the curve fitting analyses of the Mo K-edge EXAFS data for Mo/SiO2 and Mo/Al2O3. 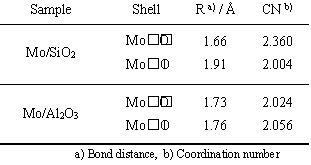
The diffuse reflectance UV-vis spectra of Mo/SiO2 and Mo/Al2O3 exhibited broad absorption bands at around 220-240 and 260-280 nm (data not shown) which have been assigned to the ligand to metal charge transfer absorption bands of the tetrahedrally coordinated Mo6+-oxide species [15]. Furthermore, these catalysts do not exhibit any absorption band of MoO3 in the wavelengths region above 350 nm, showing that these Mo6+-oxide species exist in a highly dispersed state as supported by the Mo K-edge XAFS results. The reaction mechanism of the selective oxidation of CO in the presence of H2 was also investigated in detail. Under UV irradiation at around 300 nm, Mo/SiO2 exhibited photoluminescence at around 450 nm at 298 K attributed to the radiative decay process from the charge transfer excited triplet state of the isolated tetrahedral Mo6+-oxide species ((Mo5+-O-)*) [16, 17]. The absorption and emission processes can be represented by the following equation (Eq. 1). 
Figure 4 (A-C) show the effect of the addition of CO, O2 and H2 on the photoluminescence spectra of Mo/SiO2. Photoluminescence is efficiently quenched by the addition of CO, O2 and H2, accompanying the decrease in the photoluminescence lifetime, indicating that the Mo6+-oxide species dynamically interact with CO, O2 and H2 in its charge transfer excited triplet state. The evacuation of the added quencher molecules after the quenching of the photoluminescence led to almost a complete recovery of the original photoluminescence yield for O2 and H2, while only a partial recovery of the photoluminescence yield could be observed for CO, indicating that an irreversible reaction proceeds between CO and the charge transfer excited triplet state of Mo6+-oxide species ((Mo5+-O-)*). On the other hand, Mo/Al2O3 exhibited quite weak photoluminescence (data not shown) as compared to that of Mo/SiO2. From the results of XAFS measurements, the weak photoluminescence intensity of Mo/Al2O3 can be explained by the efficient thermal deactivation from photoexcited Mo6+-oxide species to support, which is enhanced by the strong interaction between Mo6+-oxide species and Al2O3 as suggested by XAFS results. The efficient thermal deactivation process of photoexcited state of Mo6+-oxide species on Al2O3 dramatically decrease catalytic reaction rate on Mo/Al2O3. 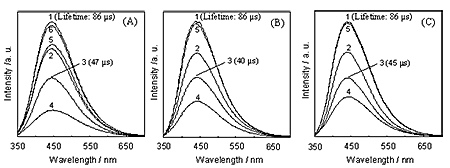
Figure 4. Effect of the addition of CO (A), O2 (B) and H2 (C) on the photoluminescence spectrum of the Mo/SiO2 at 298 K (λex=300 nm). A: 1) 0.0; 2) 2.96; 3) 12.8; 4) 110.6 ppm; 5) degassed for 1 h; 6) addition of O2 and then degassed for 1 h after 5), B: 1) 0.0; 2) 7.90; 3) 38.5; 4) 131.3 ppm; 5) degassed for 1 h, C: 1) 0.0; 2) 952.6; 3) 4240; 4) 13700 ppm; 5) degassed for 1 h. Figure 5 shows the Stern-Volmer plots for the quenching of the photoluminescence of Mo/SiO2. The Stern-Volmer equation (Eq. 2) can be obtained for the quenching of the photoluminescence with the quencher molecules by applying steady-state treatment [18], as follows: Φ0/Φ = 1+τ0 kq [Q] (2) where Φ0 and Φ show the yields of the photoluminescence in both the absence and presence of quencher molecules, respectively, and τ0, kq and [Q] are the lifetimes of the charge transfer excited triplet state of the Mo6+-oxide species ((Mo5+-O-)*) in the absence of quencher molecules, the quenching rate constant and the concentration of the quencher molecules, respectively. The absolute quenching rate constants (kq) for each gas determined by the slope of the Stern-Volmer plots increases in the following order: H2 << O2 < CO, indicating that CO interacts most efficiently with the charge transfer excited triplet state of Mo6+-oxide species ((Mo5+-O-)*), while the interaction is weak in the case of H2, as shown in Figure 5. 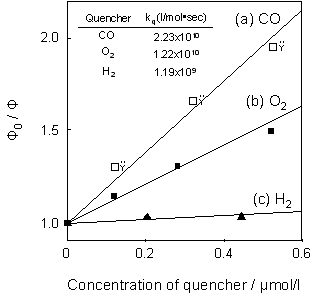
Figure 5. Stern-Volmer plots of the Φ0 / Φ values for the yields of the photoluminescence versus the concentration of the quencher such as CO (a), O2 (b) and H2 (c). (Lifetime of Mo/SiO2 under vacuum: 86 μsec at 298 K.) The reaction mechanism was also investigated by FT-IR measurements. As shown in Figure 6 (a), UV irradiation of Mo/SiO2 in the presence of CO (13130 ppm) led to the appearance of FT-IR peaks due to the dicarbonyl (Mo4+(CO)2; 2127, 2077 cm-1) and monocarbonyl (Mo4+(CO); 2043 cm-1) species, indicating that Mo6+-oxide species are photoreduced to Mo4+ carbonyl species [19]. However, the addition of O2 on this system led to the complete disappearance of these FT-IR peaks (Figure 6(b)), indicating the regeneration of tetrahedrally coordinated Mo6+-oxide species by the addition of O2. This redox cycle is also confirmed by the fact that the photoluminescence yield of the photoreduced sample is recoverd to the original level by the addition of O2 and followed its evacuation (Figure 3 (A)). According to the above characterization, the catalytic reaction cycles in the photocatalytic oxidation of CO on the tetrahedrally coordinated Mo6+-oxide species on SiO2 under UV irradiation could be considered as follows (Eq. 3-5). 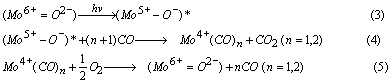
That is, initially, tetrahedral Mo6+-oxide species ((Mo6+=O2-)) on SiO2 are photoexcited into its charge transfer excited triplet state ((Mo5+-O-)*) reacting selectively with CO to form CO2 and photoreduced into Mo4+ carbonyl species. Then the Mo4+ carbonyl species are oxidized by O2 and the original Mo6+-oxide species are generated. Thus, it can be concluded that the redox cycles of Mo6+-oxide species play a significant role in the selective oxidation reaction of CO in the presence of H2. As mentioned above, the CO selectivity for this reaction on TiO2 (89%) is lower than that of Mo/SiO2 (99%). It has been reported that photoformed oxygen species (O-, O2- and O3-) play an important role in the photocatalytic oxidation reaction on TiO2 [20]. The strong and nonselective oxidation ability of these photoformed oxygen species may lead to the low CO selectivity of the reaction on TiO2. A more detailed study of the mechanisms behind the selective photocatalytic oxidation of CO is now underway. 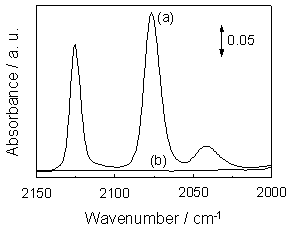
Figure 6. FT-IR spectra observed under UV light irradiation of Mo/SiO2 in the presence of CO (a); and the effect of the addition of O2 in the dark on the FT-IR spectra of CO adsorbed species formed on the pre-photoreduced Mo/SiO2 with CO (b). All spectra were recorded as the difference in the spectrum before and after UV light irradiation. Conclusions UV-vis, photoluminescence and XAFS measurements revealed that Mo6+-oxide species are present in a highly dispersed state in a tetrahedral symmetry on SiO2 and Al2O3. The selective photocatalytic oxidation of CO in the presence of H2 proceeded efficiently on the tetrahedrally coordinated Mo6+-oxides species on SiO2 with high CO conversion (~100%) and selectivity (99%) under UV light irradiation for 180 min. It was found that this reaction hardly proceeded on the tetrahedrally coordinated Mo6+-oxides species on Al2O3, since the strong interaction of Mo6+-oxide species with Al2O3 results in the enhanced thermal deactivation process of the photoexcited Mo6+-oxide species on Al2O3. Furthermore, in-situ spectroscopic investigations revealed that the reaction is closely related to the high reactivity of the charge transfer excited triplet state of Mo6+-oxide species ((Mo5+-O-)*) on SiO2 to oxidize CO into CO2, as well as the high reactivity of the photoreduced Mo4+-oxide species with O2 to produce the original Mo6+-oxide species. Acknowledgements The authors wish to thank NSERC, Canada for financial support for this work. References 1. S. Gottesfeld and J. Pafford, “A New Approach to the Problem of Carbon Monoxide Poisoning in Fuel Cells Operating at Low Temperatures”, J. Electrochem. Soc., 135 (1988) 2651-2652. 2. H.-F. Oetjen, V.M. Schmidt, U. Stimming and F. Trila, “Performance Data of a Proton Exchange Membrane Fuel Cell using H2/CO as Fuel Gas”, J. Electrochem. Soc., 143 (1996) 3838-3842. 3. M. Watanabe, H. Uchida, K. Ohkubo and H. Igarashi, “Hydrogen Purification for Fuel Cells: Selective Oxidation of Carbon Monoxide on Pt-Fe/Zeolite Catalysts”, Appl. Catal. A, 46 (2003) 595-600. 4. M. Shou, K. Tanaka, K. Yoshioka, Y. Moro-oka and S. Nagano, “New Catalyst for Selective Oxidation of CO in Excess H2 Designing of the Active Catalyst Having Different Optimum Temperature”, Catal. Today, 90 (2004) 255-261. 5. M. Echigo and T. Tabata, “A Study of CO Removal on an Activated Ru Catalyst for Polymer Electrolyte Fuel Cell Applications”, Appl. Catal. A, 251 (2003) 157-166. 6. S. Dzwigaj, M. Matsuoka, M. Anpo and M. Che, “Effect of the Addition of Propane and Distortion of Tetrahedral Vanadium(V) Species in VSi Beta Zeolites on the Photodecomposition of NO”, Res. Chem. Intermed., 29 (2003) 665-680. 7. H. Yamashita, K. Yoshizawa, M. Ariyuki, S. Higashimoto, M. Che and M. Anpo, “Photocatalytic Reactions on Chromium Containing Mesoporous Silica Molecular Sieves (Cr-HMS) under Visible Light Irradiation: Decomposition of NO and Partial Oxidation of Propane” Chem. Commun., (2001) 435-436. 8. H. Yamashita, S. Ohshiro, K. Kida, K. Yoshizawa and M. Anpo, “Visible-light-responsive Photocatalytic Reaction on Tetrahedrally-coordinated Chromium Oxide Moieties Loaded on ZSM-5 Zeolites and HMS Mesoporous Silica: Partial Oxidation of Propane”, Res Chem. Intermed., 29 (2003) 881-890. 9. R. Tsumura, S. Higashimoto, M. Matsuoka, H. Yamashita, M. Che and M. Anpo, “Investigation on the Photoluminescence Properties of Mo-MCM-41 and the Photocatalytic Decomposition of NO in the Presence of CO”, Catal. Lett., 68 (2000) 101-103. 10. I.R. Subbotina, B.N. Shelimov, V.B. Kazansky, A.A. Lisachenko, M. Che and S. Coluccia, “Selective Photocatalytic Reduction of Nitric Oxide by Carbon Monoxide Over Silica-supported Molybdenum Oxide Catalysts”, J. Catal., 184 (1999) 390-395. 11. T. Kamegawa, R. Takeuchi, M. Matsuoka and M. Anpo, “Photocatalytic Oxidation of CO with Various Oxidants by Mo Oxide Species Highly Dispersed on SiO2 at 293 K”, Catal. Today, 111 (2006) 248-253. 12. R. Radhakrishnan, C. Reed, S.T. Oyama, M. Seman, J.N. Kondo, K. Domen, Y. Ohminami and K. Asakura, “Variability in the Structure of Supported MoO3 Catalysts: Studies using Raman and X-ray Absorption Spectroscopy with Ab Initio Calculations”, J. Phys. Chem. B, 105 (2001) 8519-8530. 13. S. Takenaka, T. Tanaka, T. Funabiki and S. Yoshida, “Structure of Molybdenum Species in Silica-supported Molybdenum Oxide and Alkali-ion-modified Silica-supported Molybdenum Oxide”, J. Phys. Chem. B, 102 (1998) 2960-2969. 14. Y. Iwasawa, “Chemical Design Surfaces for Active Solid Catalysts”, Adv. Catal., 35 (1987) 187-264. 15. N. Giordano, J.C.J. Bart, A. Vaghi, A. Castellan and G. Martinotti, “Structure and Catalytic Activity of MoO3∙Al2O3 Systems I. Solid-state Properties of Oxidized Catalysts”, J. Catal., 36 (1975) 81-92. 16. M. Anpo and M. Che, “Applications of Photoluminescence Techniques to the Characterization of Solid Surfaces in Relation to Adsorption, Catalysis, and Photocatalysis”, Adv. Catal., 44 (1999) 119-257. 17. M. Anpo, M. Kondo, S. Coluccia, C. Louis and M. Che, “Application of Dynamic Photoluminescence Spectroscopy to the Study of the Active Surface Sites on Supported Mo/SiO2 Catalysts. Features of Anchored and Impregnated Catalysts”, J. Am. Chem. Soc., 111 (1989) 8791-8799. 18. N.J.Turro in “Modern Molecular Photochemistry” (N.J. Turro, Ed.), Benjamin/Cummings, New York, (1978). 19. C.C. Williams and J.G. Ekerdt, “Infrared Spectroscopic Characterization of Molybdenum Carbonyl Species Formed by Ultraviolet Photoreduction of Silica-supported Mo(VI) in Carbon Monoxide”, J. Phys. Chem., 97 (1993) 6843-6852. 20. S. Sato and T. Kadowaki, “Photocatalytic Activities of Metal Oxide Semiconductors for Oxygen Isotope Exchange and Oxidation Reactions”, J. Catal., 106 (1987) 295-300. Contact Details |