Image Credit: Yosuke Sumiya, Yu Harabuchi, Yuuya Nagata, Satoshi Maeda. JACS Au. April 22, 2022.
Predicting the formula for the desired product molecule with no other information than just the molecule itself might be a useful tool for speeding up reaction development. Previously, the Maeda group devised a computational approach for forecasting single-step reactions in this manner.
When it comes to multi-step reactions, conversely, the number of alternative reaction routes skyrockets — a phenomenon known as combinatorial explosion. Calculation costs are prohibitively expensive because of the fast-growing complexity.
To get over this limitation, scientists devised an algorithm that lowers the number of paths that need to be examined by removing less plausible paths at each phase of the process. A kinetic analysis approach examines how efficiently each path produces the target molecule after evaluating all potential paths for one step back in the reaction.
Reaction pathways that do not produce the target molecule over a pre-determined proportion are judged insignificant and are not pursued further.
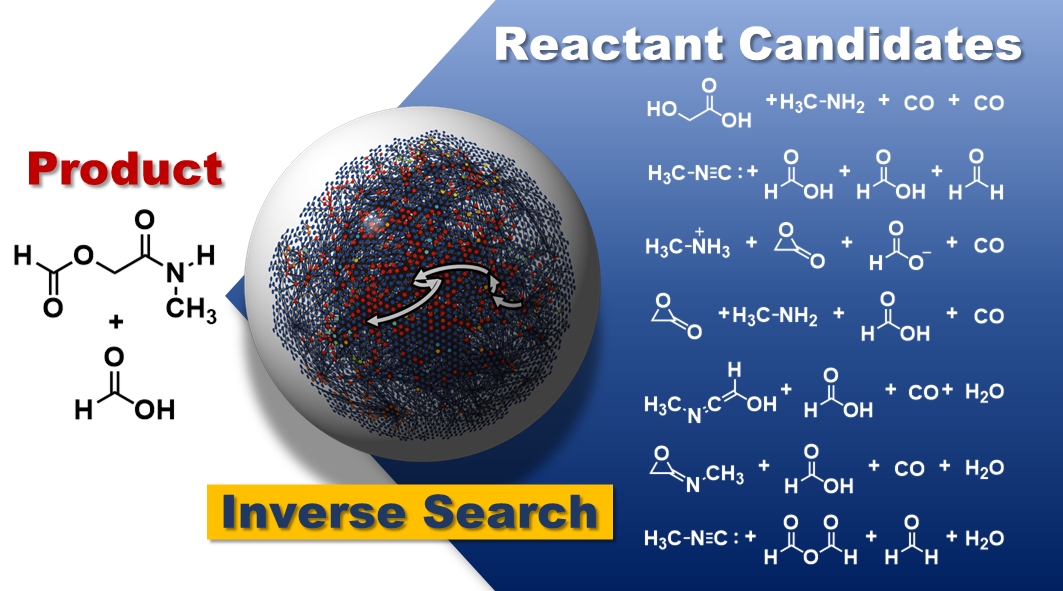
In a multi-step reaction, this cycle of exploring, assessing and eliminating reaction routes is continued for each step backward, reducing the combinatorial explosion that would otherwise occur and making multi-step reactions more calculable. Previous approaches could only predict single-step reactions, but this new method could predict reactions with more than six stages, which is a significant improvement.
The approach was tried on two well-known multi-step reactions, the Strecker and Passerini reactions, as a solid evidence experiment. For each reaction, thousands of potential starting materials were provided, which were narrowed down to the most viable options based on durability and product yield.
The well-known beginning materials for each reaction were among the offered candidates, validating the technique’s capacity to find experimentally feasible starting materials when only given the intended product molecule.
Although additional research is needed to predict even greater and more complicated systems, experts believe that this advance in multi-step process management will speed up the discovery of new chemical reactions.
This work provides a unique approach, as it is the first time performing reverse predictions of multi-step reactions using quantum chemical computations is possible without using any knowledge or data about the reaction. We expect this technique will enable the discovery of entirely unimagined chemical transformations, in which case there is little knowledge or experimental data to use.
Satoshi Maeda, Professor, Hokkaido University
Journal Reference:
Sumiya, Y., et al. (2022) Quantum Chemical Calculations to Trace Back Reaction Paths for the Prediction of Reactants. JACS Au. doi.org/10.1021/jacsau.2c00157.