Machine learning (ML) is being extensively used in materials science. It is supposed that a model developed by ML could show the common trend of the data and thus reflect the association between structure and property, which can be applied to several of the compounds. So, by training ML models with current databases, vital properties of compounds can be predicted before laborious experiments or calculations, which will significantly accelerate the process of new materials design.
 The machine learning results. (a) The scatter plot, and (b) the histogram of errors and the kernel density estimation of the probability density function. Red points and regions correspond to structures with prediction error larger than 2 eV. (Image credit: ©Science China Press)
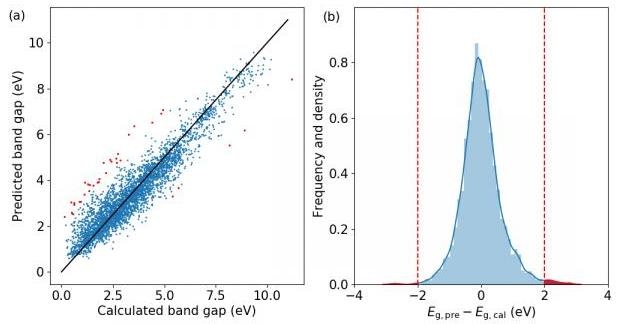
The machine learning results. (a) The scatter plot, and (b) the histogram of errors and the kernel density estimation of the probability density function. Red points and regions correspond to structures with prediction error larger than 2 eV. (Image credit: ©Science China Press)
While extremely useful, these models do not immediately exhibit the rules and physics behind the relationship between structure and property. Plus, regardless of their decent general performance, there will constantly be a few exceptions where ML models fail to deliver accurate predictions. Very frequently, it is these exceptions that offer little insights about the underlying physics, and pave the way to new frontiers in science.
A study team led by Prof. Feng Pan, the founding dean of the School of Advanced Materials, Peking University Shenzhen Graduate School, has now demonstrated that these models are valuable not only when they are successful in predicting properties correctly, but also when they falter. In their research, a model is constructed to predict the HSE band gaps of compounds based on their atomic structures only, according to a high-throughput calculation database put together by the school themselves. The R2 of the model is 0.89, comparable with analogous works. They then strained out those structures with prediction error larger than 2 eV and analyzed them carefully. A number of structures with odd structure units, or displaying other abnormities with similar compounds, like comparatively large band gaps or being in different phases. Among these odd structures, AgO2F raises significant interest and a thorough analysis is given. It is found that Ag3+ and O22 coexist in this compound, and although Ag ions are in square planar coordination, there is only limited hybridization between orbitals of Ag and O. States closer to the band edges are mostly contributed by O-2p orbitals and the band gap is a lot smaller than other compounds with Ag3+ ions. This offers a new instance for anionic redox property, a talking point in the study of Li-excess electrode materials. These results show how uncommon structures can be discovered from exceptions in ML, which can help the team to explore new physics and unique structural units from current databases.